

Авторы: А.С. Бавыкин 1, М.А. Волкова 2
1Институт молекулярной биологии РАН, 2Российский Онкологический научный центр им. Н.Н. Блохина РАМН, Москва
Современная интенсивная терапия острых нелимфобластных лейкозов (ОНЛЛ) нередко проводится на грани возможностей организма больного перенести ее. В первую очередь это относится к больным старше 60 лет, которые, составляя не менее половины всех больных ОНЛЛ, часто не получают интенсивной терапии и не включаются в рандомизированные исследования эффективности различных лечебных режимов из-за опасности развития угрожающих жизни осложнений. В то же время использование лекарственных средств, действие которых направлено на устранение молекулярного дефекта, лежащего в основе развития лейкоза, как показывает опыт терапии острого промиелоцитарного лейкоза полностью трансретиноевой кислотой (ATRA), может проводиться независимо от возраста и давать одинаково высокие результаты во всех возрастных группах [1–3].
В течение последних двух десятилетий ведутся активные исследования молекулярных изменений при лейкозах и разработка лекарственных средств, направленных на устранение молекулярных дефектов. Использование выявленных молекулярных маркеров в качестве прогностических факторов позволяет разделить пациентов на группы, требующие терапии различной интенсивности. Успехи цитогенетики и изучение молекулярных дефектов, обусловленных хромосомными аберрациями, позволили разделить ОНЛЛ на 3 группы. Первая – группа со сбалансированными хромосомными аберрациями, т.е. с аберрациями, при которых не происходит потери генетического материала. Эта группа представлена в основном реципрокными транслокациями с образованием химерных генов, в результате чего нарушается функция генов, кодирующих транскрипционные факторы, играющие ключевую роль в гемопоэзе. Больные с аберрациями этой группы при современной терапии имеют благоприятный прогноз [4]. Вторая – группа с несбалансированными хромосомными аберрациями, т.е. с аберрациями, при которых происходит потеря генетического материала, главным образом с делециями – потерей части или утратой целых хромосом. Плохой прогноз у больных с хромосомными аберрациями этого типа позволяет предполагать в патогенезе ОНЛЛ этой группы роль антионкогенов, утрачиваемых в результате хромосомных потерь [5]. Третью группу, в которую входят около половины всех больных ОНЛЛ, составляют больные с нормальным кариотипом. Отсутствие видимых цитогенетических нарушений затрудняет поиски молекулярных дефектов, поэтому патогенез лейкозов данной группы остается неясным. Больные с нормальным кариотипом традиционно относятся к группе с промежуточным прогнозом. Однако результаты применения различных лечебных программ показывают, что внутри этой группы существуют прогностически различные подгруппы.
В последние годы выявлены некоторые типы нарушений клеточного генома, которые не обусловлены или, по крайней мере, не всегда обусловлены видимыми хромосомными аберрациями, но вовлекают гены, играющие важную роль в гемопоэзе и в патогенезе лейкозов.
Первой аномалией, обнаруженной при ОНЛЛ с нормальным кариотипом, была реаранжировка гена MLL (ген смешанной лейкемической линии – mixed lineage leukemia) [6]. Ген MLL локализован на хромосоме 11 в районе 11q23. Транслокации, вовлекающие район 11q23 и различные другие хромосомы (всего обнаружено более 30 различных транслокаций с участием 11q23), встречаются при ОНЛЛ и при острых лимфобластных лейкозах (ОЛЛ) с экспрессией миелоидных маркеров. Изучение изменений гена MLL у больных с различными аберрациями, вовлекающими район 11q23, показало, что при трисомии 11 с очень высокой частотой встречается частичное тандемное удвоение MLL [7, 8]. Вскоре было обнаружено, что такая же реаранжировка гена MLL встречается у 6-8% больных ОНЛЛ с нормальным кариотипом [8, 9]. Исследование больших групп больных показало, что частичное тандемное удвоение гена MLL никогда не встречается при транслокациях, ассоциированных с хорошим прогнозом – t(8;21), t(15;17), inv 16. Среди больных ОНЛЛ с нормальным кариотипом обнаружение реаранжировки MLL предвещает плохой прогноз [9, 10]. Вторая генная реаранжировка, обнаруженная у больных ОНЛЛ с нормальным кариотипом, – реаранжировка гена FLT3.
Рецептор FLT 3: структура и функции
Рецептор FLT3, или CD 135, относится к семейству клеточных белков, которые совмещают в себе функции рецептора и внутриклеточной тирозинкиназы. Из членов этого семейства наиболее известны такие рецепторы, как рецептор к фактору роста эндотелия сосудов (VEGFR 2), рецептор к фактору роста стволовых клеток (c-kit) и а- и в-рецепторы к фактору роста, продуцируемому тромбоцитами (PDGFR). Белок FLT3 кодируется геном flt3, который расположен на длинном плече хромосомы 13 (13q12.2). Ген flt3 состоит из 24 экзонов, длина его транскрипционной РНК составляет 2979 пар оснований. Молекулярная масса FLT3-тирозинкиназы – 112804 Д при длине в 993 аминокислоты.Молекулу FLT3 можно условно разделить на две функциональные единицы: рецептор и тирозиновую киназу, связанные друг с другом трансмембранным доменом (ТМ). Рецепторная часть расположена на клеточной мембране, она имеет характерную структуру – состоит из пяти иммуноглобулиноподобных доменов, которые могут связываться с лигандом (рис. 1). Внутриклеточная часть состоит из подмембранного домена (JM- юкстамембранный) и двух каталитических (киназных) доменов (TK1 и TK2).
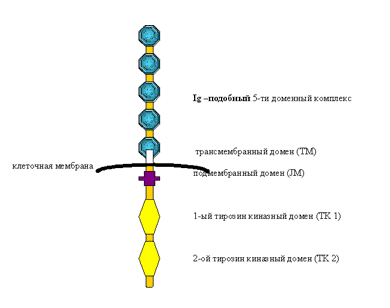
Рис. 1. Схема строения одиночной молекулы рецептора FLT3
Экспрессия FLT3-тирозинкиназы обнаружена преимущественно на стволовых CD34+-кроветворных клетках и на гемопоэтических клетках- предшественниках в костном мозге, на лимфоцитах тимуса и лимфоузлов, менее выраженная экспрессия обнаружена на клетках плаценты, головного мозга, мозжечка и на половых клетках [11, 12].
Лиганд для FLT3 (FL) представляет собой пока еще не вполне охарактеризованную молекулу, которую по ее свойствам относят к семейству цитокинов. FL состоит из 235 аминокислот и представляет собой трансмембранный белок, который может переходить в растворимую форму во внеклеточном пространстве и взаимодействовать со своим рецептором. FL продуцируется клетками костномозгового микроокружения, включая фибробласты, а также гемопоэтическими клетками миелоидного и B- и T-клеточного лимфоидных рядов. FL является ростовым фактором для стволовых клеток и миелоидных предшественников и способствует пролиферации CD34+-клеток в культуре и in vivo в опытах на животных [13–17]. Как и некоторые другие лиганды (например, фактор Стила – лиганд c-kit), FL не может самостоятельно эффективно индуцировать пролиферацию нормальных миелоидных и лимфоидных предшественников, он действует совместно с другими гемопоэтическими ростовыми факторами и интерлейкинами [18, 19]. Поскольку FLT3-киназа экспрессирована на тех же CD34+-клетках, на которых определяется высокий уровень экспрессии c-kit (CD117), их лиганды (FL и фактор Стила), очевидно, действуют в данной популяции клеток синергично [20]. В культуре клеток и в опытах на мышах была показана способность FL совместно с гранулоцитарно-макрофагальным фактором и интерлейкином-4 индуцировать дифференцировку дендритных клеток, а совместно с интерлейкином-15 – увеличивать число натуральных киллеров [21–24].
В неактивном состоянии рецептор представлен в виде одиночных молекул (см. рис. 1). Взаимодействие лиганда с рецептором вызывает димеризацию двух одиночных молекул (рис. 2, а), и в этом состоянии рецептор становится активным. Этот процесс сопровождается фосфорилированием тирозиновых остатков тирозинкиназного домена TK 2 (рис. 2, б). Эти аминокислотные остатки формируют так называемые зоны связывания для внутриклеточных белков – Src-киназ, что приводит к запуску каскада реакций (по MAP-киназному пути для FLT3), итогом которых является экспрессия генов, отвечающих за пролиферацию и рост клеток.
Механизм экспрессии FLT3 в норме и при гемобластозах
Нарушение процесса рецепторной регуляции внутриклеточных структур является основным звеном в патогенезе острого миелолейкоза. Мутации FLT3 приводят к независимой от лиганда димеризации и неконтролируемой активации рецептора (см. рис. 2). Процессы в норме показаны на рис. 2, а красными линиями и стрелками в серединной части рисунка, синими стрелками – при патологии (см. рис. 2, б; помимо нормальной передачи сигнала через Src-белки включается дополнительная передача пролиферативного сигнала через STAT5). Особое внимание следует обратить на так называемую А (активационную) петлю, которая служит для связывания внутриклеточных киназ. Процесс связывания в норме зависит от двух основных факторов: активированное (димеризованное) состояние рецептора и его сродство к Src-киназам (адапторные белки). В неактивном состоянии рецептора (одиночные молекулы FLT3) отсутствует контакт с Src-киназами. При мутации в А-петле (см. рис. 2, б) изменяется аминокислотная последовательность в ней, и это может быть чревато сразу несколькими аномальными явлениями. Во-первых, А-петля в результате произошедшей мутации, как правило, переходит в состояние постоянной активации и проводит сигнал вне зависимости от контакта FLT3-рецептора с лигандом. В этом случае запускаемый каскад реакций становится более продолжительным, чем в норме, что может привести к неконтролируемой стимуляции экспрессии соответствующих генов, направленных на пролиферацию и ангиогенез. Во-вторых, А-петля становится доступной не только для Src-киназ, но и для других активационных факторов, например STAT 5. Эти факторы, в свою очередь, запускают активацию уже «своих» генов, что, в частности, может приводить к блокированию клеточного апоптоза.
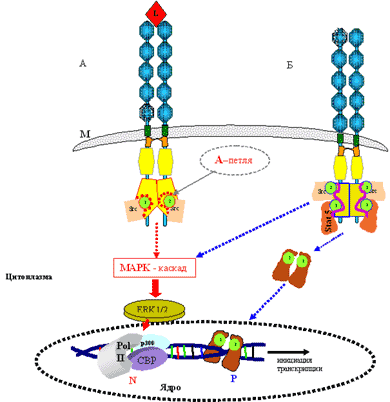
Рис. 2. Схематическое изображение активации FLT3 L – лиганд, М – цитоплазматическая мембрана, N – активация рецептора в норме, P – активация рецептора в патологии.
Исследование экспрессии FLT3 на бластных клетках при гемобластозах показало ее высокий уровень у 70–100% больных ОНЛЛ и у 87-100% больных В- и Т-клеточными острым лимфобластным лейкозом (ОЛЛ) [25–27]. Экспрессия FLT3 не была обнаружена на клетках крови и костного мозга больных хроническим миелолейкозом, но очень высокий уровень экспрессии обнаружен при бластном кризе независимо от его иммунологического варианта [28]. Уровень экспрессии FLT3 бластными клетками миелоидной, В- и Т-лимфоидных лейкемических линий значительно превосходит уровень ее экспрессии клетками нормального костного мозга. Это позволило предположить, что экспрессия FLT3 может играть роль в пролиферации и выживании лейкемических клеток [26]. В одном из исследований было показано, что в 40 из 110 изученных клеточных линий человеческих лейкозов и лимфом на лейкемических клетках была обнаружена одновременно экспрессия FLT3 и FL. Это позволяет предполагать возможность существования аутокринного механизма пролиферации лейкемических клеток [29].
Мутации гена FLT3
Мутации в юкстамембранном домене. Впервые мутации гена FLT3 при ОНЛЛ были обнаружены в 1996 г. M. Nakao и соавт. [30]. Изучая с помощью полимеразной цепной реакции структуру гена FLT3 у больных ОНЛЛ, они неожиданно обнаружили удлинение фрагментов РНК юкстамембранного (подмембранного) домена у 5 из 30 больных. Исследуя ДНК генома FLT3 этих больных, они исключили возможность удлинения домена за счет альтернативного сплайсинга нуклеотидов и показали, что во всех случаях удлинение было обусловлено тандемным удвоением в последовательности нуклеотидов, иногда со вставками добавочных нуклеотидов. Удвоенный участок варьировал по величине и локализации у разных больных, но всегда находился в пределах экзонов 14 и 15 юкстамембранного домена FLT3. Поскольку образующиеся при этом последовательности нуклеотидов всегда находились в пределах рамки считывания и не нарушали ее, было очевидно, что эти изменения не препятствуют продукции функционально активной FLT3-тирозинкиназы. Последующие многочисленные исследования показали, что обнаруженное внутреннее тандемное удвоение нуклеотидов в ДНК юкстамембранного домена FLT3 (FLT3/ITD – internal tandem duplication) является наиболее частой мутацией у больных ОНЛЛ, в том числе у больных с нормальным кариотипом. Частота FLT3/ITD при ОНЛЛ, по данным разных исследований, составляет от 13,2 до 32% у взрослых больных [31, 32]. Эта мутация была обнаружена у 3% больных с миелодиспластическими синдромами [33, 34], у отдельных больных ОЛЛ, чаще с бифенотипическим иммунофенотипом [33, 35] и не обнаружена при хроническом миелолейкозе, хроническом лимфолейкозе, неходжкинских лимфомах [34] и у здоровых доноров в клетках костного мозга и пуповинной крови, имеющих физиологически высокую экспрессию FLT3 [36, 37]. Исследование роли произошедшей мутации на культуре клеток Cos7 с трансфектным геном FLT3/ITD показало, что в них происходит независимое от лиганда фосфорилирование рецептора при любой длине и локализации удвоенного участка и что при этом одновременно происходит фосфорилирование FLT3 дикого типа на другом аллеле в тех же клетках [38]. Трансфекция FLT3/ITD в линии клеток, рост которых в норме происходит только в присутствии ростовых факторов, вызывала пролиферацию клеток без добавления этих факторов. При этом в клетках активизировались сигнальные пути с участием STAT5 и RAS/MAP-киназ [23, 24].
На сегодняшний день считается, что процесс димеризации в норме пространственно ограничивается структурным строением подмембранного домена (JM). В случае ITD структура домена JM нарушается таким образом, что процесс сближения двух рецепторных молекул оказывается в значительной степени облегченным и может происходить и без наличия лиганда.
Тандемные удвоения в юкстамембранном домене FLT3 у больных ОНЛЛ с этим дефектом значительно различаются по длине удвоенного участка – от 3 до более чем 400 пар оснований [39]. Имеются также различия в локализации тандемного участка, однако в него всегда оказывается включенным тирозин в 589, 591, 597 или 599-й позиции в молекуле FLT3 [40]. Поскольку данная мутация не всегда оказывается простым тандемным удвоением, а часто сопровождается добавочными включениями нуклеотидов, нередко вместо термина «внутреннее тандемное удвоение FLT3» (FLT3/ITD) в настоящее время применяется термин «мутация длины FLT3» (FLT3 length mutation – FLT3-LM). Мутации в киназном домене FLT3 В 2001 г. двумя независимыми группами исследователей была описана еще одна разновидность мутаций гена FLT3 – точечные нуклеотидные замены [41, 42]. Наиболее часто встречающейся и поэтому наиболее известной в настоящее время является мутация во втором киназном домене (ТК2), в нуклеотидном триплете 835, который кодирует аминокислоту аспарагин. Чаще всего происходит замена аспарагиновой кислоты на тирозин, иногда на валин, гистидин или другую аминокислоту. Помимо этой мутации при ОНЛЛ могут встречаться и другие замены. Преимущественно они локализуются в области между 835 и 842-м триплетами. Наиболее известной является замена метионина на изолейцин в 836-м триплете [41, 42]. Указанные триплеты кодируют аминокислоты, которые являются структурными элементами А-петли домена TK2.
В клетках линии Cos7 с трансфекцией FLT3 с мутациями в киназном домене происходит фосфорилирование тирозина без участия лиганда, а в клеточной линии 32D, для роста которой в обычных условиях необходим интерлейкин-3, – пролиферация без добавления интерлейкина [41]. Эти исследования показывают, что мутации в киназном домене гена FLT3 в клеточной культуре вызывают такой же, хотя и менеее выраженный, пролиферативный эффект, как FLT3/ITD. Частота мутаций в киназном домене FLT3 при ОНЛЛ, по данным разных исследований, составляет 3–7%. Наличие всего одной точечной мутации достаточно для изменения конфигурации А-петли, которая становится доступной не только для Src-киназ, но и для белков STAT 5, которые принадлежат семейству STAT (Signal Transducers and Activators of Transcription) – факторов активаторов транскрипции (см. рис. 2, б). Известно, что избыточная экспрессия белков семейства STAT может провоцировать онкогенез, поскольку они регулируют экспрессию генов, которые кодируют регуляторы клеточного цикла – циклины D1/D2, С-myc, индуктор ангиогенеза – VEGF и ингибитор апоптоза – Bcl-xL. STAT относятся к цитоплазматическим факторам и имеют в своем составе ТК- и ДНК-связывающиеся домены. После связывания с тирозинкиназными доменами рецептора тирозиновые остатки STAT5 фосфорилируются, происходит димеризация двух отдельных молекул и активированный STAT 5-комплекс мигрирует в ядро к промоторным зонам соответствующих генов.
Частота встречаемости FLT3/ITD при различных вариантах ОНЛЛ
Исследования, проведенные на больших группах больных, показали, что частота мутаций не связана с возрастом и полом, хотя в отдельных работах указывается более частая встречаемость мутации FLT3/ITD у женщин [43]. Наиболее часто у взрослых больных FLT3/ITD встречается при остром промиелоцитарном лейкозе (ОПЛ), особенно часто при его вариантной форме – М3v [44, 45]. На втором месте по частоте обнаружения FLT3/ITD находится острый монобластный лейкоз, его вариант М5b. Реже данная мутация обнаруживается при М1-, М2- и М4-вариантах ОНЛЛ, а при М0, М6 и М7 – лишь у отдельных больных. Особенно редко FLT3/ITD встречается у больных с t(8;21), аберрациями 11q23, del 5q и комплексными изменениями кариотипа. Все различия в частоте встречаемости, по данным имеющихся исследований, оказались высокодостоверными (р<0,006–0,0001) [39, 40]. Почти так же часто, как при ОПЛ, FLT3/ITD обнаруживается у больных ОНЛЛ с нормальным кариотипом.
У детей частота FLT3/ITD при ОНЛЛ примерно такая же, как у взрослых, хотя имеются некоторые отличия в частоте данной мутации при различных вариантах ОНЛЛ. Как и у взрослых, более чем 50% больных детей с FLT3/ITD имеют нормальный кариотип [46, 47]. Однако в отличие от взрослых основную группу больных с FLT3/ITD составляют больные с М1- и М2-вариантами ОНЛЛ – на их долю приходится 59% среди всех обследованных больных с данной мутацией, хотя так же, как у взрослых, FLT3/ITD очень редко встречалась при М2 с t(8;21) – лишь у 1 из 25 обследованных больных [46]. В отличие от взрослых FLT3/ITD редко обнаруживается у детей с М3. FLT3/ITD не обнаружена ни у кого из детей с частыми при детском ОНЛЛ аберрациями 11q23 (47). Ch. Zwaan и соавт. [47] обнаружили FLT3/ITD достоверно чаще у более старших детей: медиана возраста больных с мутацией составила 13,4 года, без указанной мутации – 8,8 года (р<0,001). В отличие от взрослых мутация чаще встречалась у мальчиков [47]. В табл. 1 приведены данные о частоте FLT3/ITD при ОНЛЛ.
Табл. 1 Частота FLT3/ITD при различных вариантах ОНЛЛ
| Автор | Чис-ло боль-ных | Об-щая час-тота, % | % боль-ных с норма-льным карио-типом | Вариант, % | |||||||
| М0 | М1 | М2 | М3 | М4 | М5 | М6 | М7 | ||||
| ВЗРОСЛЫЕ | |||||||||||
| P.D. Kottaridis [40] | 854 | 27 | 0 | 36 | 25 | 7 | 0 | ||||
| Ch. Thiede [44] | 979 | 20,4 | достов. чаще | 31 | |||||||
| S. Schnittger [39] | 1003 | 23,5 | 70,5 | 8,7 | 35,3 M3v-65 |
0 | 0 | ||||
| F. Kuchen-bauer [45] | 170 (М3) | 38,4 | 24,1 M3v-64,5 bcr-62,3 |
M5a-6,4 M5b-34,4 |
|||||||
| ДЕТИ | |||||||||||
| N.Y. Lacayo [46] | 81 | 30 | >50 | 59 | 20 | ||||||
| Ch.M. Zwaan [47] | 234 | 11,5 | |||||||||
Во всех исследованиях у взрослых больных с FLT3/ITD были достоверно выше количество лейкоцитов и процент бластных клеток в крови и костном мозге. Это подтверждает возможную роль FLT3 в пролиферации бластных клеток. В то же время у детей с FLT3/ITD не обнаружено ни характерного для взрослых более высокого лейкоцитоза, ни более высокого процента бластных клеток в крови и костном мозге.
Частота мутаций в киназном домене FLT3(FLT3/TKD) у взрослых больных ОНЛЛ Мутации в киназном домене FLT3 – FLT3/TKD встречаются значительно реже, чем FLT3/ITD. При ОНЛЛ, по данным разных исследований, частота FLT3/TKD колеблется от 6,4 до 7,7%. Эти мутации также встречаются чаще у больных с нормальным кариотипом [41–43]. FLT3/TKD обнаружена у 3,4% больных миелодиспластическими синдромами [41]. По данным немецкого кооперированного исследования, включившего 2535 больных ОНЛЛ, мутации в тирозинкиназном домене FLT3 (FLT3/TKD) обнаружены у 8,6% больных с нормальным кариотипом и у 4,7% больных с хромосомными аберрациями. Наиболее часто мутации в киназном домене встречались у больных с М4 эозинофильным вариантом и inv16 и при М5-варианте, особенно при M5b [48]. Частота FLT3/TKD при М3, по разным данным, составляет 8,8–19% и так же, как FLT3/ITD, чаще встречается при М3v. Как и внутренние тандемные удвоения, мутации в киназном домене почти не встречаются при М6- и М7-вариантах ОНЛЛ [41, 43, 48]. Частота FLT3/TKD при ОНЛЛ у детей достоверно неизвестна в связи с редкостью у них этого вида острого лейкоза и единичными проведенными исследованиями.
Мутации в киназном домене FLT3 у детей с ОЛЛ Оба вида рассмотренных мутаций гена FLT3 крайне редки у взрослых больных с ОЛЛ – они обнаружены лишь у 2–3% пациентов [33, 41]. В то же время мутации в киназном домене обнаружены с высокой частотой у детей с ОЛЛ, при этом, в отличие от ОНЛЛ, гораздо чаще у больных с хромосомными аберрациями, главным образом, у больных с гиперплоидией или с реаранжировкой гена MLL. По данным T. Taketani и соавт. [49], ни у одного из 162 детей с ОЛЛ не было обнаружено FLT3/ITD. В то же время мутации в киназном домене обнаружены у 18,2% больных детей с реаранжировкой гена MLL и у 21,5% – с гиперплоидией. FLT3/TKD чаще встречалась у маленьких детей: у 5,4% детей старше 1 года и у 16% в возрасте до 1 года. Не было обнаружено корреляции частоты мутаций в киназном домене с полом ребенка, высотой лейкоцитоза, числом тромбоцитов, наличием гепато- и спленомегалии и нейролейкемии [49]. Эти данные подтверждены S. Armstrong и соавт. [50], которые обнаружили мутации в киназном домене FLT3 у 25% детей с ОЛЛ с гиперплоидией. По их данным, мутации гораздо реже встречались при реаранжировке MLL [50]. В табл. 2 суммированы данные о частоте FLT3/TKD. По имеющимся данным, мутации в киназном домене FLT3 не влияли на результаты терапии детей.
Таблица 2. Частота FLT3/TKD при различных вариантах острых лейкозов
| Автор | Чис-ло боль-ных | Об-щая час-тота , % | Норма-льный карио-тип, % | Хромо-сомные аберра-ции | Варианты | |||||||
| M0 | M1 | M2 | M3 | M4 | M5 | M6 | M7 | |||||
| Взрослые – ОНЛЛ | ||||||||||||
| Ch. Thiede [48] | 2235 | 6,5 | 8,6 <0,001 |
4,7p <0,001 |
3,5 | 7,1 | 4,2 | 8,8 | 10,2 | 11,4 | 0 | 0 |
| ДЕТИ – ОЛЛ | ||||||||||||
| T. Taketani [49] | 162 | Гипер-плоидия, % 21,5 |
Реаранжировка MLL, % 18,2 |
|||||||||
| S. Armstrong [50] | 71 | 25 | <3 | |||||||||
Прогностическое значение FLT3/ITD- и TKD-мутаций при ОНЛЛ
Прогноз при FLT3/ITD. Оценке прогностической роли FLT3/ITD при ОНЛЛ посвящено большое количество исследований. В большинстве из них сравнительный анализ продемонстрировал негативное влияние данной мутации на течение и результаты терапии. Частота ремиссий при современной терапии у больных с FLT3/ITD в большинстве исследований достоверно не отличалась от частоты ремиссий у больных без данной мутации, однако безрецидивная выживаемость в большинстве исследований у больных с FLT3/ITD оказалась достоверно короче, что ухудшало конечные результаты терапии. Так, в британском кооперированном исследовании, включившем 854 больных ОНЛЛ, частота ремиссий у больных без мутации составила 84%, у больных с FLT3/ITD –78%, что не являлось статистически достоверной разницей. В то же время различия в 5-летней безрецидивной выживаемости (46% у больных без мутации, 30% у больных с FLT3/ITD) и общей выживаемости (44 и 32% соответственно) были высокодостоверными (p<0,001) [37]. В исследовании F. Abu-Duhier и соавт. [31], включившем 106 больных, при отсутствии достоверных различий в частоте ремиссий были существенные различия в выживаемости у больных с FLT3/ITD (12,8 мес в среднем) и без мутации (29,1 мес, р=0,0002). Все пациенты с FLT3/ITD, кроме одного, умерли в течение 18 мес от начала заболевания [31]. Аналогичные данные получены в кооперированном исследовании из Франции. Частота ремиссий была одинаковой в группе без мутации FLT3 (80%) и с FLT3/ITD (78%), однако общая выживаемость оказалась различной за счет различий в эффективности лечения рецидивов: 3-летняя выживаемость после лечения рецидивов составила 27% в группе без мутации и 0% в группе с FLT3/ITD [51]. Это положение подтверждается и для больных с ОПЛ, у 96–99% которых при современной терапии достигается ремиссия. При рецидиве у больных с FLT3/ITD удается получить лишь очень кратковременные ремиссии, большинство больных погибают в короткие сроки после рецидива [52].
Интересные данные получены в немецких кооперированных исследованиях, включивших 1003 больных. Все больные первоначально получили интенсивную терапию (двойная индукция TAD9/TAD9 или TAD9/HAM или HAM/HAM). Частота ремиссий составила 70,3% в группе с FLT3/ITD и 70,4% в группе без мутации, не было и достоверной разницы в общей выживаемости, но безрецидивная выживаемость была достоверно хуже в группе с FLT3/ITD [39]. Эти данные показывают, что интенсивная терапия может изменить конечные результаты, поскольку отсутствие разницы в общей выживаемости между больными двух групп в этом исследовании, очевидно, было достигнуто за счет успешного лечения рецидивов, однако негативное влияние FLT3/ITD сохраняется. Это положение подтверждается британским кооперированным исследованием, в котором проанализированы результаты трансплантации стволовых кроветворных клеток у 311 больных. Результаты индукционной терапии были одинаковыми в группе больных с FLT3/ITD и без данной мутации – в обеих группах получено 85% ремиссий. После достижения ремиссии 141 больной получил аутотрансплантацию стволовых кроветворных клеток, 170 – аллотрансплантацию от сиблинга. Разницы в числе ауто- и аллотрансплантаций между группами больных с FLT3/ITD и без нее не было, однако частота рецидивов за 10 лет наблюдения достоверно различалась: 70% в группе с FLT3/ITD, 51% – в группе без тандемного удвоения (p<0,001). Это отразилось на общей выживаемости: 10 лет прожили 27% больных в группе с FLT3/ITD, 39% – в группе без нее. Авторы делают вывод, что даже включение аллотрансплантации в лечебную программу не дает возможности преодолеть негативное влияние FLT3/ITD [53].
У детей влияние FLT3/ITD оказалось даже более универсальным, чем у взрослых: оно касается не только безрецидивной и общей выживаемости, но и частоты ремиссий. В американском кооперированном исследовании при лечении 91 ребенка в результате индукционной терапии было получено 40% ремиссий у больных ОНЛЛ с FLT3/ITD и 74% у больных без данной мутации, а 8-летняя безрецидивная выживаемость составила 7 и 44% соответственно (p<0,002) [54]. Эти данные подтверждены исследованием, включившем 234 ребенка: частота ремиссий составила 70% в группе с FLT3/ITD и 88% в группе без нее, а 5-летняя безрецидивная выживаемость 29 и 46% соответственно (p<0,0046) [47].
Значение сохранения нормального аллеля FLT3 при FLT3/ITD мутации. В последние годы появились исследования, показавшие, что на прогноз больных ОНЛЛ влияет не только наличие FLT3/ITD, но и соотношение мутировавших рецепторов и нормальных рецепторов – рецепторов «дикого» типа FLT3/WT. Было обнаружено, что у больных с FLT3/ITD мутация исчезает при достижении ремиссии, но в рецидиве появляется вновь, при этом в ряде случаев происходит уменьшение или полная утрата рецепторов FLT3/WT. Это показывает, что утрата нормального аллеля, по всей вероятности, играет роль в прогрессировании заболевания [39]. S. Whitman и соавт. [55] детально проследили динамику гематологических изменений и влияние на прогноз постепенной утраты нормального аллеля FLT3. Ими было показано, что по мере утраты FLT3/WT нарастают лейкоцитоз и число бластных клеток в крови, оба показателя были наиболее высокими у больных с отсутствием FLT3/WT. Оценка прогностического значения отсутствия FLT3/WT показала, что ремиссии в этой группе достигаются несколько реже, но различия недостоверны: 86% ремиссий в группе без FLT3/ITD, 79% – с FLT3/ITD и сохранением нормального аллеля, 75% у больных без FLT3/WT. Однако продолжительность ремиссии у больных с отсутствием FLT3/WT оказалась достоверно и значительно короче: медиана продолжительности ремиссии составила 52, 24 и 4 мес соответственно, а процент больных, проживших 12 мес, – 71, 51 и 17 (p<0,0017). У 6 из 8 больных с FLT3/ITD и отсутствием нормального аллеля была получена ремиссия после индукционной терапии, однако у 5 из 6 рецидив развился в течение ближайших 3 мес. Один год прожили 74% больных без мутаций FLT3, 65% с FLT3/ITD и лишь 13% в группе без FLT3/WT (p<0,0005) [55]. Аналогичные результаты получены Ch. Thiede и соавт. [44]. При интенсивной терапии (двойная индукция и высокодозная консолидация) не было разницы в частоте ремиссий у больных без мутаций FLT3 и с FLT3/ITD (71,2 и 66,8% соответственно), но соотношение FLT3/ITD и FLT3/WT имело решающее значение для безрецидивной и общей выживаемости. Если это соотношение превышало 2, результаты были исключительно плохими: у 14 из 15 больных этой группы развился быстрый рецидив, и все 14 умерли в течение года после достижения ремиссии с медианой выживаемости 8 мес. Ремиссия сохранялась лишь у больного, получившего аллогенную трансплантацию [44]. Таким образом, можно сделать вывод, что больные ОНЛЛ с FLT3/ITD и утратой нормального аллеля имеют исключительно плохой прогноз. Несколькими исследованиями показана также неблагоприятная роль наличия тандемных удвоений в нескольких местах гена FLT3 у одного и того же больного. P. Kottaridis и соавт. [37], обследовав 224 больных ОНЛЛ с FLT3/ITD, установили, что у 23% имеется не одно, а 2–4 тандемных удвоения. У этих больных были наиболее высокие лейкоцитоз и процент бластных клеток в крови и костном мозге. Многофакторный анализ показал, что количество мутаций является независимым неблагоприятным прогностическим признаком для частоты ремиссий, безрецидивной и общей выживаемости [37]. Такие же данные получены Ch. Thiede и соавт. [44].
Прогноз при FLT3/TKD. Анализ прогностического значения FLT3/TKD показал различные результаты в разных исследованиях. По данным Ch. Thiede и соавт. [44], эти мутации не оказывают влияния на частоту ремиссий, но снижают общую и безрецидивную выживаемость. В то же время S. Frohling и соавт. [32] и Y. Yamamoto и соавт. [41] не обнаружили влияния этих мутаций на показатели общей и безрецидивной выживаемости. Тем не менее два метаанализа, включившие 1160 и 2535 больных ОНЛЛ (249 из них с мутациями в киназном домене), показали отсутствие влияния FLT3/TKD на частоту ремиссий, но отрицательное влияние на общую и безрецидивную выживаемость [48, 56].
Сочетание мутаций гена FLT3 с мутациями других генов
В отдельных случаях, главным образом у пожилых больных, обнаруживается сочетание мутаций гена FLT3 и других генов, чаще всего гена RAS, участвующего в передаче пролиферативного сигнала, и гена ТР53, являющегося супрессором опухолевого роста. Мутации RAS выявлены примерно у 20% больных ОНЛЛ. В большинстве исследований не обнаружено влияния этих мутаций на результаты терапии [57, 58]. Мутации ТР53 обнаруживаются примерно у 15% больных ОНЛЛ, преимущественно у больных старших возрастных групп. При этих мутациях отмечено снижение общей и безрецидивной выживаемости [59]. D. Stirewalt и соавт. [60], исследовав 140 больных, показали, что сочетание мутаций указанных генов встречается редко: в их наблюдениях мутации FLT3 обнаружены у 29% больных, RAS – у 19%, ТР53 – у 9%, сочетание мутаций FLT3 и RAS обнаружено лишь у 2 больных, сочетания мутаций FLT3 и ТР53 не обнаружено. По всей вероятности, в индуцировании мутаций FLT3 и ТР53 участвуют разные механизмы, поскольку мутации ТР53 в 90% случаев встречаются у больных с неблагоприятными хромосомными аберрациями (изменения хромосомы 7 и отсутствие хромосомы 5), в то время как мутации FLT3 практически не встречаются у больных с этими хромосомными аберрациями [60].
Профиль экспрессии генов у больных с мутациями FLT3
Использование микрочипов для определения различий в экспрессии генов при разных вариантах острых лейкозов показало, что при ОНЛЛ без мутаций FLT3, с мутациями FLT3/ITD и FLT3/TKD экспрессируются различные гены. Обнаружено наибольшее прогностическое значение экспрессии двух генов: RUNX3, действующего как опухолевый супрессор, и ATRX, участвующего в метилировании ДНК. Оказалось, что по соотношению экспрессии этих генов больных ОНЛЛ с мутациями FLT3 можно разделить на 3 прогностические группы. В группе с низким соотношением RUNX3/ATRX (менее 2) 3 года без рецидива прожили 62% больных, в группе с соотношением от 2 до 10 – 37%, при соотношении более 10 – 0%. Общая трехлетняя выживаемость в группе с низким соотношением составила 70%, в группе с высоким – только 7% [46].
Методы выявления мутаций в гене FLT3
Представленные нарушения FLT3 являются очень «удобными» в отношении диагностики из-за их локализации, главным образом, в двух участках гена.
Внутренние тандемные дупликации ITD наблюдаются между 14-м и 15-м экзонами гена flt3. По разным данным, размер вставки может варьировать от 2 до 400 нуклеотидов. Расстояние между этими экзонами составляет чуть более 300 нуклеотидов, поэтому вставку можно обнаружить с помощью одной полимеразной цепной реакции (ПЦР) (рис. 3, а).
ПЦР и электрофорез в агарозном геле. Для анализа геномных нарушений FLT3 используют образцы свежей периферической крови или костного мозга, из которых выделяется ДНК и затем проводится ПЦР. После этого амплифицированные фрагменты ДНК разделяются по размеру в агарозном геле. В случае крупных ITD-вставок (более 10 нуклеотидов) можно ограничиться визуальным анализом полученного результата, так как крупные вставки видны невооруженным глазом. В норме подобранные праймеры амплифицируют фрагмент в 328 пар оснований (п.о.) (рис. 3, b1). В том случае, если вставка затрагивает оба аллельных локуса гена (что случается крайне редко), продукт ПЦР мигрирует в виде одной полосы выше 328 п.о. Как правило, наблюдается 2 полосы: 328 п.о. – что соответствует нормальном аллелю и полосы большего размера – показатель наличия ITD [25].
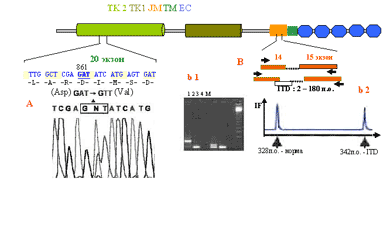
Рис. 3. ПЦР и капиллярный электрофорез.
а – секвенирование ПЦР фрагмента 20-го экзона. На сиквенсе показан смешанный пик N (в квадрате), который характеризует наличие гетерозиготной мутации в триплете 861 GАT, который в норме кодирует аспарагиновую аминокислоту. В результате замены А на Т аспарагин меняется на валин, что критическим образом меняет конформацию А-петли в домене ТК 2 при ОМЛ;
б – схема ПЦР (черными горизонтально-встречными стреками обозначены праймеры) для анализа ITD (белый блок), которая возникает в интроне (помечен пунктиром) между 14-м и 15-м (оранжевые блоки), частично затрагивая 14-й экзон. b1 – агарозный электрофорез ПЦР фрагментов, содержащих ITD различной длинны. Треки: 1 – 398 п.о; 2, 4 – норма 328 п.о.; 3 – 342 п.о. b2 – капиллярный электрофорез ПЦР фрагментов, (удобен для анализа коротких ITD). IF – интенсивность флюоресценции, L – длина фрагментов
Нередко вставка может состоять всего из нескольких нуклеотидов. Такие вставки становятся неотличимыми в агарозе. В этом случае ПЦР-фрагменты лучше всего проверять с помощью капиллярного электрофореза на секвенаторах в режиме фрагментного анализа [27]. Этот метод обладает высокой степенью разрешения и позволяет разделять фрагменты, слабо различающиеся по размеру. Метод представляет собой автоматизированный капиллярный электрофорез, в ходе которого флюоресцентно меченные ПЦР-фрагменты разделяются с высокой степенью разрешения с точностью до одного нуклеотида. На выходе из капилляра фрагменты облучаются лазером и регистрируются в виде двух пиков (интенсивности флюоресценции), отличающихся друг от друга по времени выхода из капилляра в зависимости от своей структуры и размера. Мутации, приводящие к изменению структуры А-петли, локализуются в 20-м экзоне. Наиболее значимыми являются нуклеотидные триплеты D864 (старое обозначение D835) и N870 (или -N841). Буквы даны в соответствии с названием аминокислоты и ее (или соответствующего триплета нуклеотидов) порядковым номером. Эти нуклеотидные замены также проверяются с помощью ПЦР и последующего секвенирования [61]. Анализ результатов секвенирования подробно показан на рис. 3, а. Подавляющее число мутаций находится в гетерозиготном состоянии (т.е. затрагивает только один из аллелей). В этом случае на хроматографической картинке наблюдается смешанный пик, который является результатом наложения двух сигналов – от нормального и мутантного нуклеотидов.
Ингибиторы FLT3-тирозинкиназы в терапии больных ОНЛЛ с активирующими FLT3-мутациями
Выявление роли FLT3 в гемопоэзе и значения мутаций гена FLT3 при ОНЛЛ побудило к поискам ингибиторов FLT3-тирозинкиназы. Несколько потенциальных ингибиторов было исследовано на клеточных культурах и в опытах на мышах. Два из них – СТ53518 и РКС412 – показали активность при индуцированном лейкозе мышей, удлинив латентный период до развития заболевания и увеличив выживаемость животных [62, 63].
В 2005 г. опубликованы результаты первого клинического исследования эффективности РКС412 (N-benzoylstaurosporine). Препарат не имеет узко направленного действия, он ингибирует все рецепторные тирозинкиназы данного класса. Лечение получили 20 больных с рецидивом ОНЛЛ или с рефрактерностью к ранее проведенной терапии. У 18 больных обнаружено FLT3/ITD, у 2 – мутации в киназном домене FLT3. У 14 из 18 больных, которым успешно выполнено цитогенетическое исследование, был нормальный кариотип. Препарат назначался внутрь по 75 мг 3 раза в день. Лечение пришлось отменить 8 пациентам из-за негематологической токсичности (гипербилирубинемия) или осложнений (бронхопневмония). У 14 из 20 больных после месячного лечения достигнуто более чем 50% уменьшение числа бластных клеток в крови, у 6 больных – их полное исчезновение на несколько недель и более чем 50% уменьшение в костном мозге. У 3 больных констатирована полная ремиссия, правда, при сниженной клеточности костного мозга [64].
Возможно, подобно ATRA, ингибиторы рецепторных тирозинкиназ окажутся высокоэффективными в сочетании с цитостатической терапией, что обеспечит прогресс в лечении еще одной группы больных ОНЛЛ.
Материал взят из журнала "Онкогематология", №1-2, 2006.
Литература
1. Ason N., Adachi K., Tamura J. et al. Analysis of prognostic factors in newly diagnosed acute promyelocytic leukemia treated with all-trans retinoic acid and chemotherapy. J Clin Oncol 1998;6:78–85.
2. Sanz M.A., Martin G., Rayon C. et al. A modified AIDA protocol with anthracycline-based consolidation results in high antileukemic efficacy and reduced toxicity in newly diagnosed PML/RARa –positive acute promyelocytic leukemia. Blood 1999;94: 3015–21.
3. Sanz M.A., Lo Coco F., Martin G. et al. Definition of relapse risk and role of nonanthracycline drugs for consolidation in patients with acute promyelocytic leukemia: a jont study of the PETHEMA and GIMEMA cooperative groups. Blood 2000;96:1247–53.
4. Look A.T. Oncogenic transcription factors in the human acute leukemias. Science 1997;278:1059–64.
5. Caligiuri M.A., Strout M.P., Gilliland D.G. Molecular biology of acute myeloid leukemia. Semin Oncol 1997;24:32–44.
6. Caligiuri M.A., Schichmann S.A., Strout M.P. et al. Molecular rearrangement of the ALL-1 gene in acute myeloid leukemia without cytogenetic evidence of 11q23 chromosomal translocations. Cancer Res 1994;54:372–3.
7. Tanaki N., Kaneko Y., Maseki N. et al. Trisomy 11 in chronic myelomonocytic leukemia: report of two cases and review of the literature. Cancer Genet Cytogenet 1988;30:109–17.
8. Caligiuri M.A., Strout M.P., Lawrence D. et al. Rearrangement of the ALL(MLL) in acute myeloid leukemia with normal cytogenetic Cancer Res 1998;58:55–9.
9. Dohner K., Tobis K., Ulrich R. et al. Prognostic significance of partial tandem duplication of the MLL gene in adult patients 16 to 60 years old with acute myeloid leukemia and normal cytogenetics: a study of the Acute Myeloid Leukemia Study Group Ulm J Clin Oncol 2000;20:3254–61.
10. Schnittger S., Kinkelin U., Schoch C. et al. Screening for MLL tandem duplication in 387 unselected patients with AML identify a prognostically unfavorable subset of AML. Leukemia 2000;14:796–804.
11. Agnes F., Shamoon B., Dina C. et al. Genomic structure of the downstream part of the human FLT3 gene:exon/intron structure conservation among genes encoding receptor tyrosine kinases (RTK) of subclass III. Gene 1994;145:283–8.
12. Rosnet O., Schiff C., Pebusque M.J. et al. Human FLT3/FLK2 gene: cDNA cloning and expression in hematopoietic cells. Blood 1993;82:1110–9.
13. Maroc N., Rottapel R., Rosnet O. et al. Biochemical characterization and analysis of the transforming potential of the FLT3/FLT2 receptor tyrosine kinase. Oncogene 1993;8:909–18.
14. Brasel K., Escobar S., Anderberg R. et al. Expression of the flt3 receptor and its ligand on hematopoietic cells. Leukemia 1995;9:1212–8.
15. Rasko J.E., Matcalf D., Rossner M.T. et al. The flt3/flt2 ligand: receptor distribution and action on murine haemopoietic cell survival and proliferation. Leukemia 1995; 9:2058–66.
16. Rusten L.S., Lyman S.D., Veiby O.P. et al. The FLT3 ligand is a direct and potent stimulator of the growth of primitive and commited human CD34+ bone marrow progenitor cells in vitro. Blood 1996;87:1317–25.
17. Brashem-Stein C., Flowers D.A., Bernstein I.D. Regulation of colony forming cell generation by flt3 ligand. Br J haematol 1996;94:17–22.
18. Lyman S.D., Brasel K., Roussean A.M. et al. The flt3 ligand: a hematopoietic stem cell factor whose activities are distinct from steel factor. Stem Cells 1994;12:99–107.
19. Ray R.J., Paige C.J., Furlonger C. et al. FLT3 ligand supports the differentiation of early B cell progenitors in the presence of interleukin 11 and interleukin 7. Eur J Immunol 1996;26:1504–10.
20. Rosnet O., Buhring H.J., Marchetto S. et al. Human FLT3/FLT2 receptor tyrosine kinase is expressed on the surface of normal and malignant hematopoietic cells. Leukemia 1996;10:238–48.
21. Marascovsky E., Brasel K., Teepe M. et al. Dramatic increase in the numbers of functionally mature dendric cells in FLT3 ligand-treated mice: multiple dendric cell subpopulations identified. J Exp Med 1996;184:1953–62.
22. Yu H., Fehniger T.A., Fuchshuber P. et al. FLT3 ligand promotes the generation of a distinct CD34(+) human natural killer cell progenitor that responds to interleukin 15. Blood 1998;92:3647–57.
23. Hayakawa F., Towatari M., Kiyoi H. et al. Tandem-duplicated FLT3 constitutively activates STAT5 and MAP kinase and introduces autonomous cell growth in IL-3 dependent cell lines. Oncogene 2000;19:624–31.
24. Muzuki M., Fenski R., Halfter H. et al. FLT3 mutations from patients with acute myeloid leukemia induce transformation of 32D cells mediated by the Ras and STAT pathways. Blood 2000;96:3907–14.
25. Birg F., Rosnet O., Carbuccia N. et al. The expression of FMC, kit and FLT3 in hematopoietic malignancies. Leuk Lymph 1994;13:223–7.
26. Carow C.E., Levenstein M., Kaufmann S.H. et al. Expression of the hematopoietic growth factor receptor FLT3(STK-1/FLK2) in human leukemias. Blood 1996;87:1089–96.
27. Drexler H.C. Expression of FLT3 receptor and response to FLT3 ligand by leukemic cells. Leukemia 1996;10:588–99.
28. Birg F., Courcoul M., Rosnet O. et al. Expression of the FMS/KIT-like gene FLT3 in human acute leukemias of the myeloid and lymphoid lineages. Blood 1992;80:2584–93.
29. Meierhoff G., Dehmel U., Gruss H.J. et al. Expression of FLT3 receptor and FLT3 ligand in human leukemia-lymphoma cell lines. Leukemia 1995;9:1368–72.
30. Nakao M., Yokota S., Iwai T. et al. Internal tandem duplication of the flt3 gene found in acute myeloid leukemia. Leukemia 1996;10:1911–8.
31. Abu-Duhier F.M., Goodeve A.C., Wilson G.A. et al. FLT3 internal tandem duplication mutations in adult acute myeloid leukemia define a high-risk group. Br J Haematol 2000;111:190–5.
32. Frohling S., Schlenk R.F., Breitruck J. et al. Prognostic significance of activating FLT3 mutations in younger adults (16–60 years) with acute myeloid leukemia and normal cytogenetics: a Study of the AML Study Group Ulm. Blood 2002;100:4372–80.
33. Xu F., Taki T., Yang H.W. et al. Tandem duplication of the FLT3 gene is found in acute lymphoblastic leukemia as well as acute myeloid leukemia but not in myelodysplastic syndrome or juvenile chronic myelogenous leukemia in children. Br J Haematol 1999;105:155–62.
34. Yokota S., Kiyoi H., Nakao M. et al. Internal tandem duplication of the FLT3 gene is preferentially seen in acute myeloid leukemia and myelodysplastic syndrome among various hematological malignancies. A study on a large series of patients and cell lines. Leukemia 1997;11:1605–9.
35. Nakao M., Janssen J.W., Erz D. et al. Tandem duplication of the FLT3 gene in acute lymphoblastic leukemia: a marker for the monitoring of minimal residual disease. Leukemia 2000;14:525–9.
36. Ishi E., Zaitsu M., Yhara K. et al. High expression but no internal tandem duplication of FLT3 in normal hematopoietic cells. Pediatr Hematol Oncol 1999;16:437–41.
37. Kottaridis P.D., Gale R.E., Frew M.E. et al. The presence of a FLT3 internal tandem duplication in patients with acute myeloid leukemia (AML) adds important prognostic information to cytogenetic risk group and response to the first cycle of chemotherapy: analysis of 854 patients from the United Kingdom Medical Research Council AML 10 and 12 trials. Blood 2001;98:1752–9.
38. Kiyio H., Towatari M., Yokota S. et al. Internal tandem duplication of the FLT3 gene is a novel modality of elongation mutation which causes constitutive activation of the product. Leukemia 1998;12:1333–7.
39. Schnittger S., Schoch C., Dugas M. et al. Analysis of FLT3 length mutations in 1003 patients with acute myeloid leukemia: correlation to cytogenetics, FAB-subtype, and prognosis in the AMLCG study and usefulness as a marker for the detection of minimal residual disease. Blood 2002;100:59–66.
40. Kottaridis P.D., Gale R.E., Linch D.C. FLT3 mutation and leukemia. Br J Haematol 2003;122:523–38.
41. Yamamoto Y., Kiyio H., Nacano Y. et al. Activating mutation of D835 within the activation loop of FLT3 in human hematologic malignancies. Blood 2001;97:2434–9.
42. Abu-Duhier F.M., Goodeve A.C., Wilson G.A. et al. Identification of novel FLT3 Asp 835 mutations in adult acute myeloid leukemia. Br J Haematol 2001;113:983–8.
43. Schnittger S., Boell I., Schoch C. et al. FLT3 D835/I836 point mutations in acute myeloid leukemia: correlation to cytogenetics, cytomorphology, and prognostis in 1229 patients. Blood 2002;100:329a.
44. Thiede Ch., Stendel Ch., Mohr B. et al. Analysis of FLT3-activating mutations in 979 patients with acute myelogenous leukemia: association with FAB-subtypes and identification of subgroups with poor prognosis. Blood 2002;99:4326–35.
45. Kuchenbauer F., Schoch C., Kern W. et al. Impact of FLT3 mutations and promyelocytic leukemia-breakpoint on clinical characteristics and prognosis in acute promyelocytic leukemia. Br J Haematol 2005;130:196–202.
46. Lacayo N.Y., Meshinchi S., Kinnunan P. et al. Gene expression profiles at diagnosis in de novo childhood AML patients identified FLT3 mutations with good clinical outcomes. Blood 2004;104:2646–54.
47. Zwaan Ch.M., Meshinchi S., Radich J.P. et al. FLT3 internal tandem duplication in 234 children with acute myeloid leukemia: prognostic significance and relation to cellular drug resistance. Blood 2003;102:2387–94.
48. Thiede Ch., Schnittger S., Kern W. et al. Point mutations of the FLT3-receptor tyrosine kinase in patients with acute myeloid leukemia – results of an intergroup analysis of the AML CG study and the AML96 study of the SHG. Blood 2003;102:606a, abstr. 2237.
49. Taketani T., Taki T., Sugita K. et al. FLT3 mutations in the activating loop of tyrosine kinase domain are frequently found in infant ALL with MLL rearrangements and pediatric ALL with hyperploidy. Blood 2004;103:1085–8.
50. Armstrong S.A., Mabon M.E., Silverman L.B. et al. FLT3 mutations in childhood acute lymphoblastic leukemia. Blood 2004;103:3544–6.
51. Boisel N., Cayela J.M., Predhomme C. et al. Prognostic significance of FLT3 internal tandem repeat in patients with de novo acute myeloid leukemia treated with reinforced courses of chemotherapy. Leukemia 2002;16:1699–704.
52. Callens C., Chervet S., Cayuela J.-M. et al. Prognostic implication of FLT3 and RAS mutations in patients with acute promyelocytic leukemia (APL): a retrospective study from the European APL Group. Leukemia 2005;19:1153–60.
53. Gale R.E., Hills R., Kottaridis P.D. et al. No evidence that FLT3 status should be considered as an indicator for transplantation in acute myeloid leukemia (AML): an analysis of 1135 patients, excluding acute promyelocytic leukemia, from the UK MRC AML 10 and 12 trials. Blood 2005;106:3658–65.
54. Meshinchi S., Woods W.G., Stirewalt D.L. et al. Prevalence and prognostic significance of FLT3 internal tandem duplication in pediatric acute myeloid leukemia. Blood 2001;97:89–94.
55. Whitman S.R., Archer K.J., Feng L. et al. Absence of the wild-type allele predicts poor prognosis in adult de novo acute myeloid leukemia with normal cytogenetics and the internal tandem duplication of FLT3. A Cancer and Leukemia Group B Study. Cancer Res 2001;61:7233–9.
56. Yanada M., Matsou K., Suzuki T. et al. Prognostic significance of FLT3 internal tandem duplication and tyrosine kinase domain mutations for acute myeloid leukemia: a meta-analysis. Leukemia 2005;19:1345–9.
57. Neubauer A., Dodge R.K., George S.L. et al. Prognostic importance of mutations in the ras protooncogenes in de novo acute myeloid leukemia. Blood 1994;83:1603–11.
58. Radich J.P., Kopecky K.J., Willman C.L. et al. N-ras mutations in adult de novo acute myelogenous leukemia: prevalence and clinical significance. Blood 1990;78:801–7.
59. Wattal E., Preudhomme C., Hecqet B. et al. P53 mutations are associated with resistance to chemotherapy and short survival in hematologic malignancies. Blood 1994;84:3148–57.
60. Stirewalt D.L., Kopecky K.J., Meshinchi S. et al. FLT3, RAS and TP53 mutations in elderly patients with acute myeloid leukemia. Blood 2001;97:3589–95.
61. Au W.Y., Fung A., Chim C.S. et al. FLT-3 aberrations in acute promyelocytic leukaemia: clinicopathological associations and prognostic impact. Br J Haematology 2004;125:463–9.
62. Kelly L.M., Yu J.C., Boulton C.L. et al. CT53518, a novel selective FLT3 antagonist for the treatment of acute myelogenous leukemia (AML). Cancer Cell 2002;1:421–32.
63. Levis M., Allebach J., Tse K.F. et al. A FLT3-targeted tyrosine kinase inhibitor is cytotoxic to leukemia cells in vitro and in vivo. Blood 2002;99:3885–91.
64. Stone R.M., De Angelo D.J., Klimek V. et al. Patients with acute myeloid leukemia and an activating mutation in FLT3 respond to a small-molekule FLT3 tyrosine kinase inhibitor, PKC 412. Blood 2005;105:54–60.