


Автор: Т.В. Наседкина Институт молекулярной биологии им. В.А. Энгельгардта РАН, Москва
Введение
Микрочипы как метод анализа генома получили широкое распространение в молекулярной биологии и биомедицинских исследованиях. Публикуется огромное количество статей, связанных с этой технологией, микрочипы производят многие крупные компании, а объем продаж составляет сотни миллионов долларов в год.
Идея микрочипов зародилась в конце 1980-х годов параллельно в нескольких лабораториях. В 1990 г. был опубликован патент югославских исследователей Дрманача и Чрквенякова, которые предложили иммобилизовать на двумерной поверхности анализируемую ДНК и проводить ряд последовательных гибридизаций с олигонуклеотидными пробами для определения последовательности исследуемого образца ДНК. Независимо от них российский ученый А.Д. Мирзабеков предложил определять последовательность ДНК с помощью обратного подхода, а именно иммобилизации набора коротких олигонуклеотидов, представляющих собой все возможные последовательности определенной длины, с которыми предполагалось гибридизовать анализируемый фрагмент меченой ДНК с неизвестной последовательностью. Реконструкция последовательности анализируемой ДНК должна была производиться с помощью математических методов анализа. Более подробную историческую справку можно найти в обзоре А.М. Колчинского и соавт. [1].
В дальнейшем лавинообразное развитие этих идей породило множество вариантов микрочипов, используемых в самых разнообразных областях молекулярной биологии и биомедицины [2, 3]. Хотя создано множество различных систем на основе этой технологии, можно выделить некоторые общие моменты, характерные для всех микрочипов. Прежде всего, микрочипы представляют собой устройства для проведения массового анализа по многим параметрам одновременно. Наиболее широкое применение нашли микрочипы, изготовленные на твердой поверхности. В качестве подложки используют стекло, пластик, металл, мембрана и т.п. На этой подложке могут быть иммобилизованы различные биологические макромолекулы, в первую очередь ДНК, а также РНК или белки. По устоявшейся терминологии, иммобилизуемые молекулы принято называть зондами, а те молекулы, которые находятся в исследуемом образце и подвергаются анализу, – мишенями. Как правило, зонды наносят на поверхность микрочипа в определенной последовательности; таким образом, микрочип представляет собой упорядоченную матрицу, в которой каждый элемент заранее задан и определен. В процессе гибридизации происходит специфическое взаимодействие молекул-зондов и молекулы-мишени по принципу комплементарности, а для того чтобы выявлять стабильные гибридизационные структуры и определить, с каким зондом произошло взаимодействие, молекулы-мишени предварительно метят флюоресцентным красителем. Таким образом, картина гибридизации представляет собой картину распределения флюоресцентных сигналов, наиболее ярких в точках специфического связывания зонда и мишени. В качестве примера можно рассмотреть схему строения и общий вид так называемых трехмерных микрочипов, в которых молекулы-зонды иммобилизованы не просто на поверхности стекла, а в каплях полиакриламидного геля (рис. 1). В большинстве случаев зонды синтезируют предварительно, а затем наносят на твердую подложку с помощью робота. Однако разработана также технология синтеза олигонуклеотидных зондов непосредственно на поверхности микрочипа in situ с использованием литографических масок (в микрочипах, разрабатываемых фирмой Affymetrix).
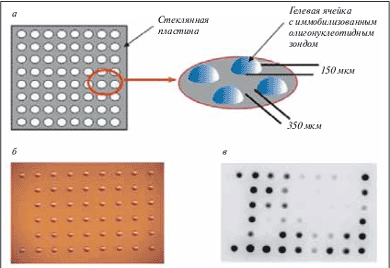
Рис. 1. Микрочип с трехмерными ячейками из полиакриламидного геля, в которых иммобилизованы олигонуклеотидные пробы.
а – строение микрочипа. Ячейки фиксированы на твердой подложке, каждая ячейка содержит индивидуальную пробу; б – фотография микрочипа в световом микроскопе; в – изображение микрочипа после гибридизации с флюоресцентно меченной мишенью. Наиболее темные точки соответствуют наиболее ярким флюоресцентным сигналам
Экспрессионные микрочипы
Одно из активно разрабатываемых направлений с применением технологии микрочипов – это исследование транскрипционных профилей при сложных заболеваниях. Хотя все клетки нашего организма обладают одной и той же переданной по наследству геномной ДНК, каждая клетка экспрессирует различные гены в виде мРНК в соответствии с типом клетки, биологическими процессами, нормальным или патологическим состоянием и т.д. Это разнообразие в профилях генной экспрессии является предметом интенсивного изучения ввиду его биологического и клинического значения. Способность технологии микрочипов анализировать экспрессию сотен и тысяч генов оказалась наиболее востребована при расшифровке такого сложного заболевания, как рак. Технология микрочипов позволяет одновременно отслеживать экспрессию десятков тысяч генов, создавая молекулярный портрет клетки [4, 5]. К наиболее значительным последствиям изучения профилей генной экспрессии можно отнести диагноз, стратификацию и определение прогноза при многих видах рака [6]. Хотя гистопатологическая оценка, дополненная цитогенетическим исследованием и анализом нескольких молекулярных маркеров, все еще является золотым стандартом в постановке диагноза и определении прогноза, работы последних лет показывают, что во многих случаях она может быть заменена определением профиля экспрессии генов. Диагноз и прогноз при раковых заболеваниях требуют совместной экспертизы нескольких специалистов-практиков, таких как онкологи, патологи и цитогенетики, кроме того, окончательные выводы могут варьировать в зависимости от методических подходов и квалификации экспертов. Микрочипы могли бы полностью заменить усилия многих специалистов, кроме того, повысить точность в постановке диагноза и определении прогноза, а также обеспечить единую стандартизированную платформу для анализа.
Для анализа экспрессии генов используют микрочипы двух типов: на основе комплементарной ДНК (кДНК) и на основе олигонуклеотидных зондов. Микрочипы на основе кДНК представляют собой фрагменты ДНК, закрепленные на поверхности стандартных микроскопических стекол или на другой твердой подложке. В олигонуклеотидных микрочипах на такой же подложке иммобилизованы олигонуклеотиды длиной 25–60 нуклеотидных оснований (н.о.). Процедура подготовки образца при проведении анализа на микрочипах представлена на рис. 2. Из клеток выделяют тотальную РНК (иногда выделяют также фракцию мРНК), далее проводят реакцию обратной транскрипции, используя комбинированный праймер, содержащий последовательность, комплементарную полиА-концевому фрагменту мРНК, и участок промотора Т7 РНК-полимеразы. Включение в состав синтезирующейся цепи кДНК последовательности промотора Т7 РНК-полимеразы позволяет в дальнейшем провести реакцию амплификации in vitro: фермент Т7 РНК-полимераза нарабатывает в пробирке множество копий РНК с каждой молекулы кДНК. Так происходит линейная амплификация исходной мРНК. Как правило, одновременно проводят мечение образующихся молекул РНК за счет использования в реакции нуклеотидов, содержащих флюоресцентную метку. В экспериментах с олигонуклеотидными микрочипами часто используют для мечения образца комплементарной РНК (кРНК) флюоресцентную метку одного типа, а уровни экспрессии генов определяют, сравнивая получаемые флюоресцентные сигналы с сигналами внутренних контрольных точек микрочипа. При работе с микрочипами на основе кДНК, как правило, в эксперименте используют 2 образца: контрольный образец метят одним флюоресцентным красителем, исследуемый образец – другим, далее их смешивают и гибридизуют с одним микрочипом. По соотношению двух разных флюоресцентных меток в каждой ячейке микрочипа судят о повышении или понижении уровня экспрессии данного гена. Независимо от технологической платформы в каждом эксперименте формируются данные, содержащие оценку уровня экспрессии десятков и сотен тысяч генов. Для обработки такого количества данных используется довольно сложный математический аппарат, в первую очередь, кластерный анализ. Анализ данных, полученных с помощью микрочипов, может производиться в сопоставлении с клиническими данными (анализ, ориентированный на проверку гипотезы, supervised analysis) или безотносительно к любой клинической характеристике пациента (независимый анализ, unsupervised analysis).
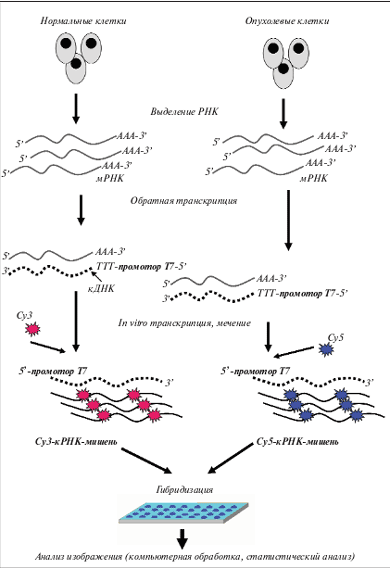
Рис. 2. Анализ на экспрессионных микрочипах при использовании двух флюоресцентных красителей. Параллельно проводят выделение РНК из нормальных и опухолевых клеток. Далее следуют этапы обратной транскрипции и in vitro транскрипции с одновременным включением флюоресцентной метки. После гибридизации в каждой точке микрочипа измеряют флюоресцентные сигналы от обоих красителей и определяют их соотношение. Таким образом определяют повышение или понижение уровня экспрессии данного гена в опухолевой клетке относительно нормальной
Анализ экспрессии генов при онкогематологических заболеваниях
Анализ профиля генной экспрессии с использованием микрочипов внес существенный вклад в наши представления о процессах, лежащих в основе онкогематологических заболеваний. Первым выдающимся примером явился анализ экспрессии генов при диффузной В-крупноклеточной лимфоме (ДККЛ) [7, 8]. ДККЛ, наиболее часто встречающийся вариант неходжкинской лимфомы, является клинически гетерогенной по ответу на терапию. Несмотря на то что большинство пациентов инициально отвечают на терапию, только около 40% остаются в длительной ремиссии. Такая гетерогенность приводит к мысли, что на самом деле это не одно, а, по крайней мере, два отличных друг от друга заболевания. В работе А.А. Alizadeh и соавт. [8] впервые была проведена стратификация пациентов с ДККЛ по экспрессии генов в клетках опухоли. Был использован микрочип на основе кДНК, содержащий фрагменты ДНК генов, участвующих в пролиферации и дифференцировке клеток лимфоидного ряда. В результате этого исследования были выявлены 2 различные по молекулярной природе формы: ДККЛ из В-клеток герминального центра (ГЦ) и ДККЛ из активированных В-клеток. При этом различные профили экспрессии генов отражали различия в скорости пролиферации, ответе на терапию и уровнях дифференцировки опухолей. При использовании одного и того же стандартного курса химиотерапии общая 5-летняя выживаемость у больных ДККЛ из В-клеток ГЦ составила 76% против 16% у больных ДККЛ из активированных В-клеток. При этом общая 5-летняя выживаемость без разделения на подгруппы составила 52%. До сих пор наиболее известным предсказателем прогноза при лимфомах служит Международный прогностический индекс (МПИ), который включает такие клинические показатели, как возраст, стадию опухоли, концентрацию лактатдегидрогеназы в сыворотке, выраженность и количество экстранодальных очагов. С использованием МПИ была выделена группа низкого риска, которая на основании анализа экспрессии генов снова дала две подгруппы с различной выживаемостью. Таким образом, было показано, что дифференциальная экспрессия генов независимо от других клинических показателей, таких как возраст пациента, стадия опухоли и т.п., может с высокой точностью определять прогноз при лимфопролиферативных заболеваниях. Эта работа вдохновила исследователей на поиски групп генов, экспрессия которых может служить независимым прогностическим фактором при стратификации пациентов по группам риска и оценке выживаемости. Одна такая попытка была сделана в работе М.А. Shipp и соавт. [9], где были выделены две категории пациентов в группе из 58 больных с диагнозом ДККЛ, которые существенно различались по 5-летней выживаемости (70 и 12%). На основе сравнительного анализа профилей генной экспрессии в двух этих группах пациентов была подобрана панель из 13 генов для использования в качестве независимого фактора прогноза.
В аналогичной работе [10] при сопоставлении клинических данных и результатов анализа экспрессии генов для 160 образцов лимфом была составлена несколько отличная панель из 17 генов, которая была успешно применена к анализу последующих 80 образцов, показав, что этот набор из 17 генов может быть использован как молекулярный предсказатель выживаемости. На основании анализа экспрессии генов были выделены 3 группы пациентов, 2 из которых соответствовали определенным ранее [8]. Параллельный анализ геномных аберраций показал, что транслокация t(14;18) с участием гена BCL-2 и амплификация гена с-REL, ранее описанные для ДККЛ, встречаются строго в одной группе, а именно у пациентов с ДККЛ из В-клеток ГЦ. Третья группа с отличающимся профилем экспрессии характеризовалась плохим прогнозом, как и ДККЛ из активированных В-клеток, но, скорее всего, не являлась однородной. Дальнейшее исследование с помощью независимых методов оценки уровня экспрессии, таких как ПЦР в реальном времени, показало большую информативность 17-генной модели, которая включала гены, относящиеся к 4 группам: гены В-клеток ГЦ, гены класса II основного комплекса гистосовместимости, гены, участвующие в развитии лимфатических узлов, и гены, участвующие в пролиферации [11]. Кроме того, были выделены всего 6 генов (LMO2, BCL6, FN1, CCND2, SCYA3, BCL2), экспрессия которых в наибольшей степени коррелировала с клиническим исходом. Фолликулярная лимфома (ФЛ) также характеризуется высокой гетерогенностью и по клиническим показателям подразделяется на опухоли с высокой и низкой пролиферативной активностью. Кроме того, ФЛ может трансформироваться в более агрессивную ДККЛ. Анализ экспрессии генов у 80 пациентов с ФЛ в сопоставлении с клиническими данными позволил выделить панель из 81 гена, пользуясь которой можно различить вялотекущую и агрессивную формы с точностью 93% [12].
Для лимфомы клеток мантийной зоны характерным является наличие хромосомной аберрации t(11;14), приводящей к повышению уровня экспрессии циклина D1 [13]. Используя микрочипы на основе кДНК, содержащие фрагменты 6386 генов, авторы проанализировали профили экспрессии для 38 случаев [14]. В последующий анализ было включено 446 генов, участвующих в апоптозе, клеточном цикле и ответственных за иммунный ответ. Наибольшее количество изменений было отмечено для сигнальных путей с участием ядерного транскрипционного фактора NFkB и фактора некроза опухоли. Предполагается, что активация NFkB-сигнального пути в ходе воспалительных процессов в организме играет важную роль в этиологии лимфопролиферативных заболеваний. В результате были выделены 26 генов, повышенный или пониженный уровень экспрессии которых можно рассматривать как прогностический фактор. С использованием этого молекулярного классификатора все пациенты были стратифицированы на 2 группы: высокого и низкого риска. В группе низкого риска, к которой были отнесены около половины всех пациентов, 5-летняя выживаемость составила 52%. В группе высокого риска ни один из пациентов не дожил до 5 лет. Медиана общей выживаемости для всех пациентов составила 27 мес.
Методологически схожие исследования были проведены на группах пациентов с острыми лейкозами. Острые лейкозы представляют собой чрезвычайно гетерогенную группу заболеваний, различающихся по происхождению, клинической картине, прогнозу заболевания и требующих различной терапии [15]. В ранних работах основное внимание было уделено поиску генов, по экспрессии которых можно было бы различать крупные нозологические формы. Например, при анализе 51 образца костного мозга детей с острыми лейкозами на микрочипах, содержащих кДНК от 4608 генов, были получены различия в профилях экспрессии между следующими группами пациентов: острый лимфобластный лейкоз (ОЛЛ) и острый нелимфобластный лейкоз (ОНЛЛ), В-клеточный лейкоз и Т-клеточный лейкоз, В-клеточный лейкоз с химерным транскриптом TEL/AML1 и лейкоз без транслокаций [16]. Таким образом, технология микрочипов оказалась в состоянии успешно стратифицировать пациентов по группам, выявляемым стандартными методами, такими как иммунофенотипирование и цитогенетический анализ. Отдельную группу лейкозов, обладающих специфическим профилем экспрессии, составляют случаи, в которых обнаружены хромосомные перестройки с участием гена MLL [17]. Ген MLL (mixed-lineage leukemia) расположен на хромосоме 11 (11q23) и часто вовлекается в хромосомные транслокации с различными генами-партнерами. Аберрации, затрагивающие регион 11q23, встречаются как при ОЛЛ, так и при ОНЛЛ. Как правило, они характеризуются плохим прогнозом, хотя на прогноз оказывает влияние также и ген-партнер, участвующий в транслокации. Было обнаружено, что для лейкозов с MLL-транслокациями в отличие от других случаев ОЛЛ и ОНЛЛ характерна повышенная экспрессия генов, ассоциированных с ранними стадиями развития клеток- предшественников гемопоэза, таких как PROML1, LMO2, FLT3, а также генов семейства HOX (так называемые гомеобоксные гены, участвующие в регуляции развития на ранних стадиях эмбриогенеза). Дальнейшие исследования подтвердили, что для всех случаев с t(11q23)/MLL независимо от гена-партнера характерна единая транскрипционная программа; в то же время внутри этой общей группы могут быть четко разделены варианты с t(11q23)/MLL при ОЛЛ и при ОНЛЛ [18–20]. Более глубокое понимание механизмов развития заболевания может играть важную роль в поиске новых подходов к лечению. Так, высокий уровень экспрессии гена FLT3, кодирующего белок семейства тирозинкиназ, позволяет рассматривать FLT3-тирозинкиназу как потенциальную терапевтическую мишень при некоторых вариантах лейкозов, в том числе и при t(11q23)/MLL [21]. Проводятся исследования по применению ингибиторов тирозинкиназы FLT3 в лечении больных лейкозами [22].
Более детальные исследования профилей генной экспрессии при ОЛЛ у детей показали, что пациенты могут быть стратифицированы по группам, соответствующим вариантам лейкозов с химерными транскриптами BCR-ABL, E2A-PBX1 и TEL-AML1, перестройками с MLL-геном, гипердиплоидным кариотипом (более 50 хромосом) и Т-клеточным лейкозом [23]. Во многом такое распределение совпало с разделением на группы на основе хромосомных аберраций. Однако в некоторых случаях классификация образца по профилю генной экспрессии может быть более точной, чем по генетической поломке. Это лучше всего иллюстрируется теми случаями, которые согласно профилю экспрессии классифицировались как носители химерного транскрипта TEL-AML1, в то время как при анализе с помощью обратной транскрипции с последующей полимеразной цепной реакцией (ОТ-ПЦР) этот химерный транскрипт обнаружен не был. В дальнейшем было показано, что каждый из этих случаев, тем не менее, имеет какие-то изменения в гене TEL. Кроме того, была выделена группа генов, по всей видимости, ассоциированных с повышенным риском развития вторичного ОНЛЛ, обусловленного терапией у пациентов с TEL-AML1 подтипом. Эти данные были проверены с использованием другого типа микрочипов, а именно олигонуклеотидных микрочипов, позволяющих анализировать около 39 000 транскриптов, и результаты стратификации пациентов на группы в целом совпали [24]. Аналогичное исследование, показывающее важность генетических изменений при ОЛЛ, было проведено на 35 образцах взрослых пациентов. Профили генной экспрессии образцов, несущих транслокации t(9;22), t(4;11), а также Т-ОЛЛ оказались очень схожими с теми, что были обнаружены у детей с ОЛЛ [25].
Т-клеточный лейкоз – агрессивная форма ОЛЛ. За последнее время ситуация была улучшена благодаря интенсификации терапии. Тем не менее известные на сегодняшний день клинические и биологические особенности Т-клеточного ОЛЛ не позволяют точно определять прогноз и выбирать подходящую по интенсивности терапию. Молекулярная природа Т-ОЛЛ была изучена в основном с помощью цитогенетики. Хромосомные аберрации при этом варианте лейкоза приводят к тому, что гены транскрипционных факторов HOX11/TLX1, TAL1/SCL, TAL2, LYL1, BHLHB1, LMO1 оказываются в непосредственной близости от сильного промотора генов иммуноглобулинов, что приводит к резкому повышению уровня их экспрессии [26]. Действительно, при анализе лейкемических клеток у детей-пациентов с Т-ОЛЛ была выявлена повышенная экспрессия онкогенных транскрипционных факторов: LYL1, HOX11L2, TAL1 [27]. На основании этих данных, были выделены группы, для которых по профилю генной экспрессии можно было определить соответствие бластных клеток определенной стадии нормального созревания лимфоцитов в тимусе. Эти стадии включали про-T-лимфоциты (корреляция с LYL1+), ранние кортикальные тимоциты (HOX11+) и поздние кортикальные тимоциты (TAL1+). Было показано, что повышенная экспрессия гена HOX11 относится к онкогенетическим событиям, ухудшающим прогноз [28]. Прогностическое значение активации гена HOX11 в дальнейшем было показано также для взрослого Т-ОЛЛ [29, 30].
При остром нелимфобластном лейкозе (ОННЛ) эффективная стратификация пациентов обычно проводится с использованием таких факторов риска, как возраст, кариотип бластных клеток, инициальный лейкоцитоз и ответ на индукционную терапию. Особенно большое значение имеют специфические хромосомные аберрации, которые используются как диагностические и прогностические маркеры [31–33]. Однако примерно в половине случаев у пациентов с ОНЛЛ обычными цитогенетическими методами изменений кариотипа не обнаруживают, что делает невозможным прогноз по данным цитогенетики. В этом случае исследование экспрессии генов открывает новые возможности в понимании механизмов возникновения ОННЛ и в определении клинического исхода в этой группе пациентов. Первые работы с применением микрочипов при анализе ОНЛЛ были направлены на то, чтобы выяснить, имеется ли соответствие между конкретным профилем генной экспрессии и специфической хромосомной транслокацией, которая определяет генотип лейкемической клетки. При исследовании 37 образцов костного мозга взрослых больных с ОНЛЛ было показано, что подтипы ОНЛЛ со специфическими хромосомными аберрациями t(8;21), t(15;17) и inv(16) могут быть независимо идентифицированы по характерному профилю генной экспрессии [34]. В другом исследовании было показано, что случаи ОННЛ с нормальным кариотипом отличаются повышенной экспрессией нескольких генов из семейства гомеобоксных (гены, участвующие в регуляции развития на ранних стадиях эмбриогенеза) [35]. Т. Yagi и соавт. [36] впервые применили анализ генной экспрессии для предсказания клинического исхода у детей с ОННЛ. Авторами был выявлен набор из 35 ассоциированных с прогнозом генов, который мог быть использован для индивидуального подбора риск-адаптированной терапии. Среди 54 пациентов с ОНЛЛ было выбрано 9 пациентов с хорошим прогнозом, которые были живы без рецидива в течение 3 лет, и 9 с плохим прогнозом, у которых или не была достигнута ремиссия, или развился рецидив в течение первого года первой ремиссии. При сравнении профилей экспрессии в двух этих группах были выбраны 135 генов, экспрессия которых характеризовалась наиболее сильными различиями. С использованием такого классификатора остальные 36 пациентов были стратифицированы на 2 группы с заметно различающейся бессобытийной выживаемостью (75 и 30%). В другом исследовании с помощью микрочипов были проанализированы 285 образцов от пациентов с ОННЛ. С применением кластерного анализа было выделено 16 групп пациентов с ОННЛ, характеризующихся определенным профилем экспрессии, включая группы с t(8;21), inv(16), t(15;17), а также группу с мутациями в гене FLT3, с моносомией 7, повышенной экспрессией гена EVI1 [37]. При анализе другой когорты из 116 взрослых пациентов с ОННЛ, из них 45 с нормальным кариотипом, было выявлено несколько новых подтипов ОННЛ [38]. Это исследование фокусируется на возможности использовать микрочипы для предсказания клинического исхода. Авторами был выбран набор из 133 генов для определения прогноза по молекулярному портрету лейкемических клеток независимо от того, имелись ли в клетках цитогенетические поломки или клетки были с нормальным кариотипом. Например, образцы с нормальным кариотипом на основании анализа экспрессии генов были разделены на 2 группы с различным прогнозом, при этом выживаемость составила 1200 дней при хорошем прогнозе и 700 дней при плохом.
Острый промиелоцитарный лейкоз (ОПЛ) ассоциирован с хромосомными транслокациями, вовлекающими рецептор ретиноевой кислоты и такие гены-партнеры, как PML и PLZF. Наибольшие успехи в лечении ОПЛ были достигнуты при применении препаратов ретиноевой кислоты (all-trans retinoic acid, ATRA), при этом участие в транслокации гена PML или PLZF определяет ретиноидчувствительный или ретиноидустойчивый вариант ОПЛ соответственно. При исследовании клеточных линий, в которых была индуцирована экспрессия или PML/RARA, или PLZF/RARA химерных транскриптов, с помощью микрочипов были выявлены гены сигнальных путей, определяющих чувствительность и устойчивость к препаратам ретиноевой кислоты. Это позволяет надеяться на использование микрочипов для определения чувствительности или устойчивости к специфическому лечению [39].
Хронический В-клеточный лимфолейкоз (В-ХЛЛ) является наиболее распространенным вариантом лейкоза у взрослых. В-ХЛЛ характеризуется моноклональной экспансией В-лимфоцитов. Это также гетерогенное заболевание по своему клиническому течению. Оно может развиваться годами и даже десятилетиями у одних больных, но в некоторых случаях быстро прогрессирует вскоре после постановки диагноза, приводя к летальному исходу [40]. Анализ соматических мутаций в вариабельных участках генов иммуноглобулинов (IgV) при В-ХЛЛ показал, что вялотекущее или прогрессирующее течение заболевания зависит от отсутствия или наличия таких мутаций [41, 42]. Была выдвинута гипотеза о том, что В-ХЛЛ в действительности может представлять собой два разных заболевания: в одном случае опухоль развивается из префолликулярных или независимых В-клеток, а в другом – из постфолликулярных В-клеток [43]. Эту гипотезу объясняет тот факт, что гены иммуноглобулинов подвергаются соматической гипермутации, проходя через центр фолликула или ГЦ. Тем более удивительным было обнаружить, что независимый (unsupervised) кластерный анализ оказался неспособным выявить различия между В-ХЛЛ с мутациями вариабельного региона (M-CLL) и В-ХЛЛ без мутаций вариабельного региона (UM-CLL) [44, 45]. В то же время по профилю экспрессии В-ХЛЛ заметно отличается от других лимфоидных опухолей и от нормальных популяций лимфоцитов [46, 47]. При использовании подхода с учетом клинических данных (supervised analysis) все же был выявлен ряд генов, по-разному экспрессирующихся при M-CLL и UM-CLL. Клиническое использование этого результата оказалось возможным, когда авторы сузили количество генов, с помощью которых удавалось дискриминировать подтипы В-ХЛЛ, до 1–3 [48]. По экспрессии гена ZAP-70 (Zeta-associated protein 70) оказалось возможным определять прогноз с точностью до 90%. Таким образом, для того чтобы отличить M-CLL и UM-CLL, достаточно простого диагностического теста, основанного на количественной ПЦР, вместо сложного анализа профилей экспрессии с помощью микрочипов или анализа последовательности ДНК вариабельного региона Ig гена. Экспрессия гена ZAP-70 коррелирует с мутационным статусом вариабельного региона Ig гена, но может также независимо определять течение заболевания и выживаемость у пациентов с В-ХЛЛ, являясь, таким образом, независимым суррогатным маркером [48, 49]. Было показано, что анализ экспрессии ZAP-70 может быть проведен на образцах периферической крови без предварительного выделения В-клеток [48]. Эти два исследования положили начало простому измерению экспрессии ZAP-70 – количественной ПЦР и проточной цитометрии. Таким образом, вывод, сделанный на основе анализа профилей генной экспрессии, уже нашел применение в прогнозировании клинического течения В-ХЛЛ [50].
Множественная миелома (ММ) является разновидностью В-клеточных злокачественных новообразований и возникает из плазматических клеток (ПК) крови. В норме эти клетки играют важную роль в иммунной системе человека. ММ характеризуется клональной экспансией ПК, неспособных выполнять защитные функции. Обычно это сопровождается повышенной выработкой моноклонального иммуноглобулина (парапротеина). Серьезным осложнением, резко ухудшающим прогноз, является выраженная инфильтрация костного мозга плазматическими клетками [51]. ММ все еще рассматривается как неизлечимое заболевание, для которого медиана выживаемости сохраняется на уровне 3 лет, несмотря на применение высокодозной химиотерапии и аутологичной трансплантации костного мозга [52]. Молекулярные механизмы болезни связывают с активной экспрессией онкогенов, которые попадают под контроль промоторов генов иммуноглобулинов в результате хромосомных аберраций. Имеются два основных момента, по которым ММ является подходящим объектом для анализа с помощью микрочипов. Во-первых, ММ является морфологически гомогенным, но клинически гетерогенным заболеванием, демонстрируя резкую разницу в выживаемости, которая может составлять от нескольких месяцев до 10 лет. Во-вторых, генетические аномалии при ММ сложны и разнообразны, так что установить корреляцию между этими аномалиями и клиническим исходом крайне затруднительно [53–55].
Первое исследование ММ с помощью микрочипов позволило сравнить экспрессию генов в ПК пациентов с ММ, моноклональной гаммопатией неустановленного значения (МГНЗ), в нормальных ПК от здоровых доноров и в клеточных линиях ММ. Были выделены 4 подгруппы больных с ММ (MM1, MM2, MM3 и MM4). Профиль экспрессии в подгруппе MM1 был близок к нормальным ПК и ПК пациентов с МГНЗ, в то время как в подгруппе ММ4 соответствовал профилю экспрессии клеточных линий ММ. Подгруппа ММ4 характеризовалась также плохим прогнозом, аномальным кариотипом и высоким уровнем ?2-микроглобулина в сыворотке крови. Среди генов с повышенным уровнем экспрессии в подгруппе ММ4 были выявлены гены FGFR3 (fibroblast growth factor receptor 3, рецептор 3 фактора роста фибробластов) и CCND1 (Cyclin D1, циклин D1). Повышенная экспрессия этих генов была обусловлена транслокациями t(4;14)(p16;q32) или t(11;14)(q13;q32) соответственно. В конечном счете был определен набор генов, позволяющих различать клетки ММ и нормальные ПК, что стало существенным вкладом в понимание молекулярной и клеточной биологии ММ [56]. Стоит отметить возможные мишени при терапии, для которых была выявлена повышенная экспрессия: это гены ABL (abl-tyrosine kinase, abl-тирозинкиназа) и CBS (cystathionine beta synthase, цистатионин бета-синтаза) [57]. Так же как и при ДККЛ [10], экспрессия генов при ММ коррелирует с различными стадиями поздней дифференцировки В-клеток в норме. Были выделены подгруппы ММ, которые, хотя и приблизительно, соответствовали по профилю экспрессии тонзилярным В-клеткам, тонзилярным ПК или костномозговым ПК [58, 59]. Наиболее интересные результаты с помощью микрочипов были получены при сравнении профилей экспрессии ПК больных ММ, имеющих и не имеющих заметного поражения костной ткани на момент постановки диагноза. Это позволило идентифицировать молекулярную составляющую остеолизиса при ММ [60]. Из 10 000 генов, экспрессия которых была измерена в этом эксперименте, были выявлены только 4 гена, экспрессия которых заметно повышалась у больных ММ с поражением костной ткани. Только один ген из этих четырех, DKK1, кодирует секретируемый белок, который функционально связан с остеобластами и поэтому выбран для дальнейшего исследования. Действительно, в последующих экспериментах было подтверждено, что продукция DKK1 клетками миеломы ассоциирована с возникновением очагов деструкции в костной ткани. Это открывает новые возможности для стратификации пациентов с ММ и поиска мишеней для дифференцированной терапии.
Обобщая эти многочисленные данные, можно сказать, что анализ профилей экспрессии генов при онкогематологических заболеваниях позволяет получить новую информацию о молекулярной природе опухоли. В практическом приложении это дает уточнение диагноза, выявление новых подтипов заболевания, предсказание ответа на терапию и клинического исхода. В конечном счете речь идет о создании принципиально новой классификации раковых заболеваний, которая позволит разрабатывать дифференцированную терапию, направленную на восстановление или нейтрализацию последствий молекулярного дефекта, вызвавшего болезнь. В одной из итоговых работ по результатам анализа образцов костного мозга 892 пациентов с различными клиническими вариантами лейкозов и 45 образцов здоровых доноров были выделены 13 подгрупп, включая пациентов с ОНЛЛ t(15;17), ОНЛЛ t(8;21), ОНЛЛ inv(16), В-ХЛЛ, пре-В-ОЛЛ с t(11q23), различными субвариантами Т-ОЛЛ и т.д. [61]. По мнению авторов, различные методы диагностики лейкозов могут быть полностью заменены таким универсальным диагностическим подходом на основе анализа экспрессии генов. Однако продвижение по этому пути происходит не так быстро, как хотелось бы, и предполагает решение еще очень многих проблем. Исследование профиля экспрессии с помощью микрочипов, содержащих пробы на тысячи и десятки тысяч генов, является слишком дорогостоящим и трудоемким для внедрения в клиническую практику. Данные, получаемые с помощью таких микрочипов, зачастую противоречивы, поскольку при использовании коммерческих продуктов разных фирм результаты могут различаться. На конечный результат исследования в значительной степени могут влиять технические различия в изготовлении микрочипов, различия в протоколах приготовления «мишеней» для гибридизации, различные алгоритмы обработки данных и т.п. Вместе с тем, для клинических целей зачастую нет необходимости в отслеживании экспрессионного профиля десятков тысяч генов, достаточно информативным оказывается анализ экспрессии нескольких десятков генов, который можно проводить с помощью более простых вариантов микрочипов. Необходимы стандартизация исследований в этой области, а также разработка недорогих и более простых в использовании подходов, которые будут доступны для каждой клинической лаборатории [62].
Анализ хромосомных перестроек с помощью микрочипов
Микрочипы являются удобным и эффективным инструментом для решения конкретных клинико-диагностических задач. Большое значение в современной диагностике лейкозов придается выявлению и идентификации хромосомных перестроек в бластных клетках. Специфические хромосомные аберрации, приводящие к образованию химерных или «слившихся» генов (fusion genes), встречаются примерно в 50% случаев ОНЛЛ и 40% случаев ОЛЛ и являются диагностическими и прогностическими маркерами различных форм заболевания, требующих различных подходов к лечению [63–65]. Анализ хромосомных перестроек с помощью традиционных цитогенетических методов трудоемок и не всегда надежен. В последнее время широкое распространение получили методы выявления самих химерных транскриптов, которые экспрессируются в бластных клетках с перестроенных последовательностей. Такую возможность дает метод ОТ-ПЦР. Однако, поскольку для выявления каждого транскрипта требуется индивидуальная реакция ПЦР со специфическими праймерами, при анализе одного пациента на наличие сразу нескольких химерных транскриптов объем работы резко возрастает.
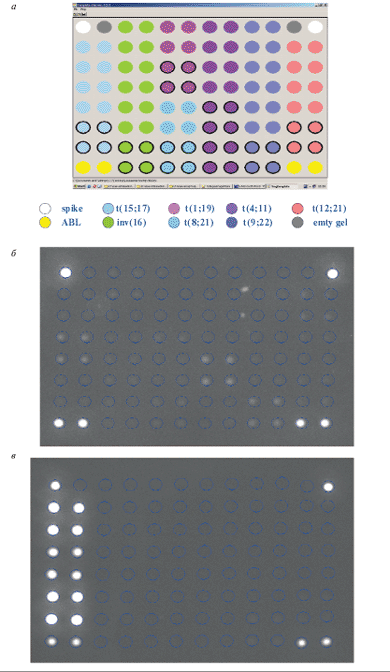
Рис. 3. Микрочип для выявления и идентификации хромосомных транслокаций.
а – схема микрочипа. Разными цветами отмечены наборы проб на различные транслокации, каждая проба представлена на микрочипе в 4-кратной повторности; б – пример гибридизационной картины, образец без транслокаций. Светятся только контрольные ячейки вверху и ячейки, содержащие пробу на ген ABL; в – пример гибридизационной картины, образец с транслокацией t(15;17). Также светятся пробы, специфичные для химерного транскрипта PML/RARA, t(15;17)
С целью упрощения и удешевления такого исследования, а также повышения специфичности анализа был разработан олигонуклеотидный микрочип, способный идентифицировать химерные транскрипты, соответствующие восьми наиболее клинически значимым транслокациям при лейкозах. Поскольку разрывы в генах при образовании транслокаций могут происходить в разных точках, одна и та же транслокация может давать начало нескольким вариантам химерных транскриптов. Набор специфических олигонуклеотидов позволяет определять 14 вариантов химерных транскриптов. На микрочипе размещены также олигонуклеотидные пробы для выявления нормального транскрипта гена ABL, экспрессирующегося во всех типах клеток. Определение ABL-транскрипта используется для контроля качества выделенной РНК и оценки эффективности всей процедуры подготовки меченого образца. Процедура анализа включает выделение РНК из клеток костного мозга или периферической крови, проведение ОТ-ПЦР в мультиплексном варианте, когда в одной пробирке проходит одновременно несколько индивидуальных ПЦР с праймерами, специфичными для разных транслокаций, гибридизацию на микрочипе и анализ изображения с помощью портативного анализатора микрочипов. На рис. 3 приведены схема микрочипа и примеры гибридизационных картин для различных случаев лейкозов. Компьютерная обработка флюоресцентных сигналов по разработанному алгоритму позволяет проводить автоматический анализ изображения. Чувствительность метода такова, что возможно обнаружение одной клетки с хромосомной перестройкой на 103 нормальных клеток, т.е. достаточна для выявления так называемой минимальной остаточной болезни. В целом достигнутая надежность выявления перестроек сравнима с таковой ОТ-ПЦР и существенно превышает надежность цитогенетических методов. С помощью разработанного микрочипа был проведен анализ образцов костного мозга 600 детей с острыми лейкозами. Полученные данные позволили определить частоту встречаемости различных транслокаций среди российских пациентов, а также оценить клиническое значение различных транслокаций (рис. 4) [66].
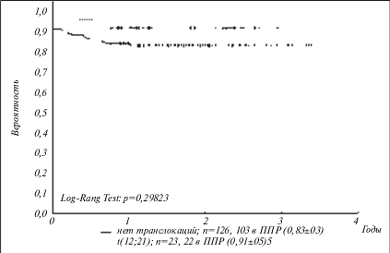
Рис. 4. Бессобытийная кривая выживаемости для пациентов с t(12;21) и без транслокаций. ППР – первичная полная ремиссия
С клинической точки зрения также важными являются перестройки с участием гена MLL (mixed lineage leukemia). Ген MLL расположен в хромосоме 11, в регионе 11q23, который часто вовлекается в хромосомные перестройки при лейкозах с образованием химерных генов. Часть химерной последовательности с 5’-конца принадлежит гену MLL, а 3’-конец – одному из генов-партнеров, которых насчитывается более 30 [67, 68]. Такое разнообразие генных аномалий с участием MLL сильно затрудняет анализ с помощью стандартных методов диагностики. В то же время задача обнаружения и идентификации хромосомных перестроек региона 11q23 особенно актуальна, так как они встречаются в 70–80% случаев ОЛЛ и 50–60% случаев ОНЛЛ у детей первого года жизни. Был разработан микрочип для анализа пяти транслокаций с участием гена MLL: t(4;11) MLL/AF4; t(9,11) MLL/AF9; t(11;19) MLL/ELL; t(11;19) MLL/ENL; dup(11) MLL/MLL. [69]. Принцип работы с MLL-микрочипом такой же, как для предыдущего варианта микрочипа на восемь транслокаций. Олигонуклеотидные пробы, расположенные в ячейках микрочипа, представляют собой участки последовательностей гена MLL и генов-партнеров, участвующих в транслокациях. При образовании транслокации ген MLL может претерпевать разрыв в различных местах (как правило, разрывы происходят после экзонов е8, е9, е10 и е11). Соответственно, на схеме микрочипа в нижней строке расположены олигонуклеотидные пробы, комплементарные участку в каждом из экзонов гена MLL. Таким образом, по картине гибридизации можно определить, какие экзоны гена MLL входят в состав химерного транскрипта, а также определить ген-партнер (рис. 5).
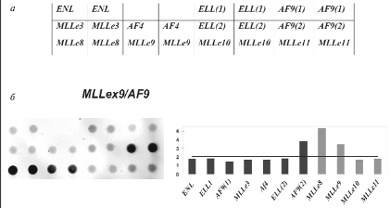
Рис. 5. Микрочип для анализа MLL-транслокаций.
а – схема микрочипа; б – пример определения транслокации t(9;11). На гибридизационной картине (слева) видны сигналы флюоресценции, на диаграмме (справа) представлены нормированные значения сигналов флюоресценции в различных ячейках микрочипа. Сигналы, превышающие пороговое значение (горизонтальная линия на диаграмме), являются специфичными и свидетельствуют о наличии химерного транскрипта MLLex9/AF9
На долю Т-клеточных лимфом приходится 15–20% всех лимфоидных опухолей. Диагностика Т-клеточных опухолей традиционно основывалась на сочетании клинических данных, гистологического анализа и иммунофенотипирования. Однако во многих случаях этого оказывается недостаточно для постановки точного диагноза, и необходимо определение Т-клеточной клональности молекулярными методами. В основе этих методов лежит анализ перестроенных локусов Т-клеточных рецепторов в популяции Т-лимфоцитов. Был разработан микрочип для определения перестроек y-цепи Т-клеточного рецептора. Микрочип содержит олигонуклеотидные зонды на все Vy- и Jy-гены, которые участвуют в перестройках y-цепи Т-клеточного рецептора. Метод также включает мультиплексную ПЦР с последующей гибридизацией на микрочипе флюоресцентно меченной «мишени». Область слияния V-J-сегментов амплифицируется только в перестроенном локусе TCR?; если y-цепь не перестроена, то V- и J-гены удалены друг от друга на значительное расстояние, что делает амплификацию невозможной. При анализе поликлональной популяции Т-лимфоцитов определяются сигналы практически от всех V?- и Jy-генов (рис. 6, а). При преобладании в образце клеток, несущих в результате реаранжировки определенную комбинацию V-J-генов, с первых циклов ПЦР-реакции начинают нарабатываться однотипные фрагменты ДНК, что и выявляется в результате гибридизации на микрочипе (рис. 6, б). Таким образом можно определять наличие моноклональных Т-клеток в клиническом образце [70].
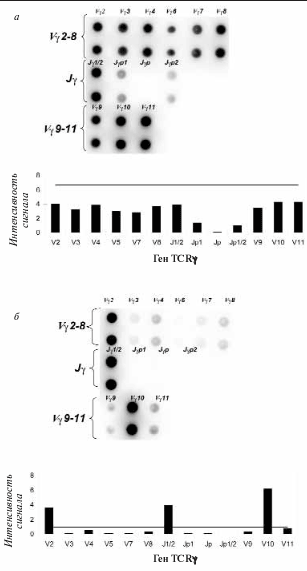
Рис. 6. Определение Т-клеточной клональности с помощью биочипа.
а – поликлональный образец здорового донора: вверху – гибридизационная картина, внизу – диаграмма распределения интенсивности флюоресцентного сигнала. Горизонтальная сплошная линия соответствует пороговому значению (ПЗ), ни один из сигналов не превышает ПЗ; б – образец больного с Т-клеточной лимфомой. Вверху – гибридизационная картина, полученная на биочипе, внизу – диаграмма распределения интенсивности флюоресцентного сигнала. Горизонтальная сплошная линия соответствует ПЗ. Сигналы от групп V?2, V?10, J?1/2 превышают ПЗ, что соответствует наличию биаллельной клональной реаранжировки V?2/ J?1/2, V?10/ J?1/2.
Преимуществами такого анализа на микрочипах являются простота использования, наглядность результата и значительное сокращение времени проведения анализа.
Анализ полиморфизма в генах системы биотрансформации и фармакогенетика
Успех в лечении онкогематологических заболеваний зависит в первую очередь от биологических свойств опухолевых клеток и чувствительности опухоли к химиотерапевтическим препаратам. Однако определенное значение имеют также характер и скорость метаболизма лекарственных средств в организме пациента. Ключевую роль в метаболизме лекарств играют ферменты системы биотрансформации, для которых характерен высокий уровень полиморфизма. Этот полиморфизм определяется однонуклеотидными заменами в генах, кодирующих соответствующие ферменты. Анализ полиморфных участков генов с помощью технологии биочипов дает важную информацию о способности организма пациента метаболизировать те или иные химиотерапевтические препараты. Эта информация может быть использована для индивидуализации терапии и корректировки дозы лекарства с тем, чтобы снизить его токсическое воздействие при лечении [71, 72].
Одним из широко применяемых в терапии лейкозов препаратом является 6-меркаптопурин (6-МП), который используется в поддерживающей терапии ОЛЛ [73]. 6-МП, а также другие тиопуриновые препараты метаболизируются с участием тиопурин-S-метилтрансферазы (ТПМТ) [74]. Активность ТПМТ наследуется по аутосомно-кодоминантному типу и демонстрирует генетический полиморфизм: около 90% людей имеют высокую активность, около 10% промежуточную активность вследствие гетерозиготности локуса гена ТПМТ, и 1 на 300 человек в популяции – низкую активность фермента (т.е. недостаточность ТПМТ), наследуя два мутантных аллеля ТПМТ [75] (рис. 7). Терапия 6-МП, 6-тиогуанином или азатиоприном в стандартных дозах у больных с недостаточностью ТПМТ приводит к развитию длительной глубокой миелосупрессии и вызывает тяжелые, нередко фатальные инфекционные осложнения [76, 77].
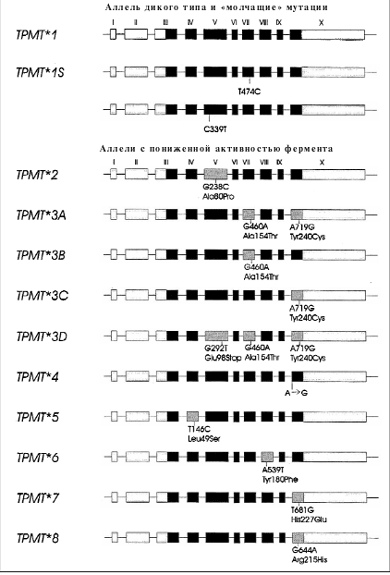
Рис. 7. Аллельные варианты гена ТПМТ
Был разработан ТПМТ-биочип для выявления наиболее распространенных мутаций в гене ТПМТ, приводящих к снижению активности фермента (рис. 8). Процесс генотипирования образцов ДНК пациентов включал выделение ДНК из лейкоцитов периферической крови, проведение мультиплексной ПЦР, гибридизацию с ТПМТ-микрочипом, регистрацию и обработку полученных результатов. Было проведено определение ТПМТ-генотипа у 280 пациентов с гемобластозами [78]. В обследованной группе больных 7,5% пациентов оказались гетерозиготными носителями мутации гена ТПМТ. Частота встречаемости и распределение мутантных аллелей гена ТПМТ в российской популяции пациентов с гемобластозами не отличались от таковой в популяции людей белой расы США и Европы [79]. Так, самым частым мутантным аллелем был ТПМТ*3А (85,7%), более редкими аллелями ТПМТ*3С (9,5%) и ТПМТ*2 (4,8%). Также ретроспективно была оценена переносимость терапии 6-МП в зависимости от ТПМТ-генотипа. Оказалось, что больные ОЛЛ с гетерозиготным генотипом ТПМТ характеризовались сниженной способностью переносить терапию 6-МП и получали достоверно более низкие дозы препарата, чем пациенты с генотипом «дикого» типа (средняя доза 6-МП в неделю – 264 против 312 мг/м2 соответственно; p = 0,04). У пациентов с гетерозиготным генотипом ТПМТ в 2 раза чаще отменяли терапию 6-МП из-за гематологической токсичности, что нарушало сроки проведения консолидации. Гетерозиготные пациенты получали больше трансфузий эритроцитов и тромбоцитов, у них чаще развивались инфекционные осложнения. Это исследование показало, что почти у 10% детей с ОЛЛ в нашей стране имеется повышенный риск тиопуриновой токсичности вследствие наследственного дефицита ТПМТ. Проспективная диагностика мутаций гена ТПМТ у больных ОЛЛ позволяет осуществлять индивидуальный подход к лечению пациентов путем редукции дозы 6-МП с целью снижения токсичности и повышения эффективности терапии в рамках установленного протокола.

Рис. 8. ТПМТ-микрочип. а – схема микрочипа, все пробы продублированы. В двух верхних рядах содержатся олигонуклеотидные пробы, комплементарные последовательностям «дикого типа», в двух нижних – соответствующие им мутантные последовательности; б и в – примеры гибридизационных картин: б – генотип «дикого типа» *1/*1, в – гетерозигота по двум позициям в гене ТПМТ, генотип *1/*3A, стрелками отмечены выявленные мутации 460A и 719G.
Генетический полиморфизм, определяемый однонуклеотидными заменами в последовательности ДНК, характерен и для других ферментов системы биотрансформации. Наиболее важными являются ферменты I фазы биотрансформации (это цитохромы семейства Р450, такие как CYP1A1, CYP2D6, CYP2C9, CYP2C19), которые активируют ксенобиотики путем формирования генотоксичных промежуточных метаболитов, и ферменты II фазы (кроме ТПМТ к ним относятся глутатион-S-трансферазы, ариламин-N-ацетилтрансферазы и др.), которые осуществляют превращение генотоксичных соединений в нетоксичные [80]. Различные полиморфные варианты ферментов, обладающие пониженной или повышенной активностью, потенциально способны модулировать ответ на терапию различными антираковыми препаратами. Это может выражаться не только в повышении токсичности лечения, но и в формировании лекарственной устойчивости или склонности к возникновению рецидивов и вторичных опухолей [81]. Был разработан биочип для анализа полиморфизма в 10 генах системы биотрансформации (рис. 9) [82]. С помощью этого биочипа был протестирован 351 образец ДНК детей, больных острыми лейкозами. Для гена CYP1A1 были выявлены распространенные сочетания аллельных вариантов *1/*1, *1/*2А, *1/*2В и *1/*4. Были определены частоты распределения CYP1A1 генотипов у пациентов с первично диагностированным лейкозом и в рецидиве заболевания. Оказалось, что среди пациентов с рецидивом ОЛЛ или ОМЛ чаще встречаются гетерозиготы по *2А аллели гена CYP1A1, чем в группе пациентов с первично диагностированным острым лейкозом (16,5 и 10% для пациентов с ОЛЛ и 19 и 10% для пациентов с ОМЛ соответственно). При этом у девочек с рецидивом острого лейкоза выявлено статистически достоверное увеличение частоты гетерозигот по *2А аллели гена CYP1A1 – более чем в 3 раза по сравнению с девочками с первично диагностированным лейкозом (относительный риск – ОР – 3,2, p < 0,05) [83]. Выявленные закономерности согласуются с данными других авторов [84, 85]. Так, в работе М. Krajinovic и соавт. [85] было обнаружено, что наличие полиморфного варианта CYP1A1*2А ассоциировано с плохим терапевтическим прогнозом у детей, больных ОЛЛ (ОР 3,1, p < 0,05).
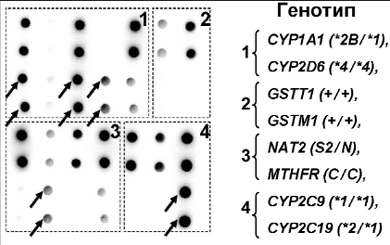
Рис. 9. Пример гибридизационной картины, полученной на биочипе для анализа полиморфизма в генах CYP1A1, CYP2D6, GSTT1, GSTM1, MTHFR, MTRR, NQO1, CYP2C9, CYP2C19 и NAT2. Стрелками указаны обнаруженные «мутантные» варианты.
В целом эти результаты указывают на то, что дальнейший прогресс в лечении онкологических заболеваний должен идти по пути индивидуализации терапии с учетом генетических характеристик пациента.
Заключение
Развитие технологии микрочипов существенно расширило область молекулярных исследований до анализа целого генома и транскриптома (совокупность всех транскриптов в клетке). Технология микрочипов используется для расшифровки сложных механизмов возникновения злокачественных новообразований, в том числе онкогематологических заболеваний. Применение микрочипов в комбинации с другими подходами должно, наконец, привести к созданию направленной дифференцированной терапии при раковых заболеваниях с учетом индивидуальных особенностей каждого пациента.
В практическом отношении применение микрочипов уже сегодня позволяет решать следующие задачи:
- точная постановка диагноза, выявление новых подтипов заболевания, уточнение классификации;
- прогнозирование течения болезни и клинического исхода, выявление генов и сигнальных путей, вовлеченных в патогенез онкогематологических заболеваний, поиск новых мишеней для направленной дифференцированной терапии;
- разработка и создание более простых и дешевых диагностических тестов, в том числе и на основе технологии микрочипов (микрочипы, содержащие пробы на десятки или сотни генов вместо десятков и сотен тысяч);
- включение микрочипов в проспективные клинические исследования, подтверждение результатов анализа на микрочипах для внесения в клинические протоколы лечения, дизайн клинических протоколов с учетом новых данных о природе заболеваний, полученных с помощью технологии микрочипов.
Материал взят из журнала "Онкогематология", №1-2, 2006.
Литература
1. Колчинский А.М., Грядунов Д.А., Лысов Ю.П. и др. Микрочипы на основе трехмерных ячеек геля: история и перспективы. Мол биол – 2004; 38(1):5–16.
2. Blohm D.H., Guiseppi-Elie A. New developments in microarray technology. Curr Opin Biotechnol 2001; 12:41–7.
3. Lockhart D.J., Winzeler E.A. Genomics, gene expression and DNA arrays. Nature 2000; 405: 827–36.
4. van Berkum N.L., Holstege F.C. DNA microarrays: raising the profile. Curr Opin Biotechnol 2001; 12: 48–52.
5. Hughes T.R., Shoemaker D.D. DNA microarrays for expression profiling. Curr Opin Chem Biol 2001; 5: 21–5.
6. Golub T.R., Slonim D.K., Tamayo P. et al. Molecular classification of cancer: class discovery and class prediction by gene expression monitoring. Science 1999; 286: 531–7.
7. Alizadeh A., Eisen M., Davis R.E. et al. The lymphochip: a specialized cDNA microarray for the genomic-scale analysis of gene expression in normal and malignant lymphocytes. Cold Spring Harb Symp Quant Biol 1999, 64: 71–8.
8. Alizadeh A.A., Eisen M.B., Davis R.E. et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature 2000; 403: 503–11.
9. Shipp M.A., Ross K.N., Tamayo P. et al. Diffuse large B-cell lymphoma outcome prediction by gene-expression profiling and supervised machine learning. Nat Med 2002; 8: 68–74.
10. Rosenwald A., Wright G., Chan W.C. et al. The use of molecular profiling to predict survival after chemotherapy for diffuse large-B-cell lymphoma. New Engl J Med 2002; 346: 1937–47.
11. Lossos I.S., Czerwinski D.K., Alizadeh A.A. et al. Prediction of survival in diffuse large-B-cell lymphoma based on the expression of six genes. New Engl J Med 2004; 350: 1828–37.
12. Glas A.M., Kersten M.J., Delahaye L.J. et al. Gene expression profiling in follicular lymphoma to assess clinical aggressiveness and to guide the choice of treatment. Blood 2005; 105(1): 301–7.
13. Hofmann W.K., de Vos S., Tsukasaki K. et al. Altered apoptosis pathways in mantle cell lymphoma detected by oligonucleotide microarray. Blood 2001; 98: 787–94.
14. Martinez N., Camacho F.I., Algara P. et al. The molecular signature of mantle cell lymphoma reveals multiple signals favoring cell survival. Cancer Res 2003; 63: 8226–32.
15. Pui C.H., Evans W.E. Acute lymphoblastic leukemia. New Engl J Med 1998; 339: 605–15.
16. Moos P.J., Raetz E.A., Carlson M.A. et al. Identification of gene expression profiles that segregate patients with childhood leukemia. Clin Cancer Res 2002; 8(10): 3118–30.
17. Armstrong S.A., Staunton J.E., Silverman L.B. et al. MLL translocations specify a distinct gene expression profile that distinguishes a unique leukemia. Nat Genet 2002; 30: 41–7.
18. Rozovskaia T., Ravid-Amir O., Tillib S. et al. Expression profiles of acute lymphoblastic and myeloblastic leukemias with ALL-1 rearrangements. Proc Natl Acad Sci USA 2003; 100(13): 7853–8.
19. Tsutsumi S., Taketani T., Nishimura K. et al. Two distinct gene expression signatures in pediatric acute lymphoblastic leukemia with MLL rearrangements. Cancer Res 2003; 63: 4882–7.
20. Kohlmann A., Schoch C., Dugas M. et al. New insights into MLL gene rearranged acute leukemias using gene expression profiling: shared pathways, lineage commitment, and partner genes. Leukemia 2005; 19(6): 953–64.
21. Armstrong S.A., Kung A.L., Mabon M.E. et al. Inhibition of FLT3 in MLL. Validation of a therapeutic target identified by gene expression based classification. Cancer Cell 2003; 3(2): 173–83.
22. Brown P., Levis M., McIntyre E. et al. Combinations of the FLT3 inhibitor CEP-701 and chemotherapy synergistically kill infant and childhood MLL-rearranged ALL cells in a sequence-dependent manner. Leukemia 2006; 20(8):1368–76.
23. Yeoh E.J., Ross M.E., Shurtleff S.A. et al. Classification, subtype discovery, and prediction of outcome in pediatric acute lymphoblastic leukemia by gene expression profiling. Cancer Cell 2002;1: 133–43.
24. Ross M.E., Zhou X., Song G. et al. Classification of pediatric acute lymphoblastic leukemia by gene expression profiling. Blood 2003; 102: 2951–9.
25. Kohlmann A., Schoch C., Schnittger S. et al. Pediatric acute lymphoblastic leukemia (ALL) gene expression signatures classify an independent cohort of adult ALL patients. Leukemia 2004; 18: 63–71.
26. Ferrando A.A., Look A.T. Gene expression profiling in T-cell acute lymphoblastic leukemia. Semin Hematol 2003; 40: 274–80.
27. Ferrando A.A., Neuberg D.S., Staunton J. et al. Gene expression signatures define novel oncogenic pathways in T cell acute lymphoblastic leukemia. Cancer Cell 2002; 1: 75–87.
28. Ballerini P., Blaise A., Busson-Le Coniat M. et al. HOX11L2 expression defines a clinical subtype of pediatric T-ALL associated with poor prognosis. Blood 2002; 100: 991–7.
29. Ferrando A.A., Neuberg D.S., Dodge R.K. et al. Prognostic importance of TLX1 (HOX11) oncogene expression in adults with T-cell acute lymphoblastic leukaemia. Lancet 2004; 363: 535–6.
30. Chiaretti S., Li X., Gentleman R. et al. Gene expression profile of adult T-cell acute lymphocytic leukemia identifies distinct subsets of patients with different response to therapy and survival. Blood 2004; 103: 2771–8. 31. Ravandi F., Kantarjian H., Giles F., Cortes J. New agents in acute myeloid leukemia and other myeloid disorders. Cancer 2004; 100: 441–54.
32. Look A.T. Oncogenic transcription factors in the human acute leukemias. Science 1997; 278: 1059–64.
33. Grimwade D., Walker H., Oliver F. et al. The importance of diagnostic cytogenetics on outcome in AML: analysis of 1612 patients entered into the MRC AML 10 trial. The Medical Research Council Adult and Children’s Leukaemia Working Parties. Blood 1998; 92: 2322–33.
34. Schoch C., Kohlmann A., Schnittger S. et al. Acute myeloid leukemias with reciprocal rearrangements can be distinguished by specific gene expression profiles. Proc Natl Acad Sci USA 2002; 99: 10008–13.
35. Debernardi S., Lillington D.M., Chaplin T. et al. Genome-wide analysis of acute myeloid leukemia with normal karyotype reveals a unique pattern of homeobox gene expression distinct from those with translocation-mediated fusion events. Genes Chromosomes Cancer 2003; 37: 149–58.
36. Yagi T., Morimoto A., Eguchi M. et al. Identification of a gene expression signature associated with pediatric AML prognosis. Blood 2003; 102: 1849–56.
37. Valk P.J., Verhaak R.G., Beijen M.A. et al. Prognostically useful gene-expression profiles in acute myeloid leukemia. New Engl J Med 2004; 350: 1617–28.
38. Bullinger L., Dohner K., Bair E. et al. Use of gene-expression profiling to identify prognostic subclasses in adult acute myeloid leukemia. New Engl J Med 2004; 350: 1605–16.
39. Park D.J., Vuong P.T., de Vos S. et al. Comparative analysis of genes regulated by PML/RAR alpha and PLZF/RAR alpha in response to retinoic acid using oligonucleotide arrays. Blood 2003; 102: 3727–36.
40. Rozman C., Montserrat E. Chronic lymphocytic leukemia. New Engl J Med 1995; 333: 1052–7. 41. Rai K.R., Sawitsky A., Cronkite E.P. et al. Clinical staging of chronic lymphocytic leukemia. Blood 1975; 46: 219–34.
42. Binet J.L., Auquier A., Dighiero G. et al. A new prognostic classification of chronic lymphocytic leukemia derived from a multivariate survival analysis. Cancer 1981; 48: 198–206.
43. Hamblin T.J., Davis Z., Gardiner A. et al. Unmutated Ig V(H) genes are associated with a more aggressive form of chronic lymphocytic leukemia. Blood 1999; 94: 1848–1854.
44. Klein U., Tu Y., Stolovitzky G.A. et al. Gene expression profiling of B cell chronic lymphocytic leukemia reveals a homogeneous phenotype related to memory B cells. J Exp Med 2001; 194: 1625–38.
45. Rosenwald A., Alizadeh A.A., Widhopf G. et al. Relation of gene expression phenotype to immunoglobulin mutation genotype in B cell chronic lymphocytic leukemia. J Exp Med 2001; 194: 1639–47.
46. Jelinek D.F., Tschumper R.C., Stolovitzky G.A. et al. Identification of a global gene expression signature of B-chronic lymphocytic leukemia. Mol Cancer Res 2003; 1: 346–61.
47. Wang J., Coombes K.R., Highsmith W.E. et al. Differences in gene expression between B-cell chronic lymphocytic leukemia and normal B cells: a meta-analysis of three microarray studies. Bioinformatics 2004; 20: 3166–78.
48. Crespo M., Bosch F., Villamor N. et al. ZAP-70 expression as a surrogate for immunoglobulin-variable-region mutations in chronic lymphocytic leukemia. New Engl J Med 2003; 348: 1764–75.
49. Wiestner A., Rosenwald A., Barry T.S. et al. ZAP-70 expression identifies a chronic lymphocytic leukemia subtype with unmutated immunoglobulin genes, inferior clinical outcome, and distinct gene expression profile. Blood 2003; 101: 4944–51.
50. Bosch F., Muntanola A., Gine E. et al. Clinical implications of ZAP-70 expression in chronic lymphocytic leukemia. Cytometry B Clin Cytom 2006; 70B(4): 214–7.
51. Kuehl W.M., Bergsagel P.L. Multiple myeloma: evolving genetic events and host interactions. Nat Rev Cancer 2002; 2: 175–87.
52. Kaufman J., Lonial S. Multiple myeloma: the role of transplant and novel treatment strategies. Semin Oncol 2004; 31: 99–105.
53. Bergsagel P.L., Kuehl W.M. Critical roles for immunoglobulin translocations and cyclin D dysregulation in multiple myeloma. Immunol Rev 2003; 194: 96–104.
54. Sawyer J.R., Waldron J.A., Jagannath S., Barlogie B. Cytogenetic findings in 200 patients with multiple myeloma. Cancer Genet Cytogenet 1995; 82:41–9.
55. Dewald G.W., Kyle R.A., Hicks G.A., Greipp P.R. The clinical significance of cytogenetic studies in 100 patients with multiple myeloma, plasma cell leukemia, or amyloidosis. Blood 1985; 66: 380–90.
56. Zhan F., Hardin J., Kordsmeier B. et al. Global gene expression profiling of multiple myeloma, monoclonal gammopathy of undetermined significance, and normal bone marrow plasma cells. Blood 2002; 99: 1745–57.
57. Claudio J.O., Masih-Khan E., Tang H. et al. A molecular compendium of genes expressed in multiple myeloma. Blood 2002; 100: 2175–86.
58. De Vos J., Thykjaer T., Tarte K. et al. Comparison of gene expression profiling between malignant and normal plasma cells with oligonucleotide arrays. Oncogene 2002; 21: 6848–57.
59. Zhan F., Tian E., Bumm K. et al. Gene expression profiling of human plasma cell differentiation and classification of multiple myeloma based on similarities to distinct stages of late-stage B-cell development. Blood 2003; 101: 1128–40.
60. Tian E., Zhan F., Walker R. et al. The role of the Wnt-signaling antagonist DKK1 in the development of osteolytic lesions in multiple myeloma. New Engl J Med 2003; 349: 2483–94.
61. Haferlach T., Kohlmann A., Schnittger S. et al. Global approach to the diagnosis of leukemia using gene expression profiling. Blood. 2005; 106(4): 1189–98.
62. Staal F., Cario G., Cazzaniga G. et al., Consensus quidelines for microarray gene expression analyses in leukemia from three European leukemia networks. Leukemia 2006; 20(8): 1385–92.
63. Pui C.H., Campana D., Evans W.E. Childhood acute lymphoblastic leukaemia – current status and future perspectives. Lancet Oncol 2001; 2: 597–607.
64. Shrappe M. Evolution of BFM trials for childhood ALL. Ann Hematol 2004; 83 (Suppl 1): 121–3.
65. Pui C.H., Relling M.V., Sandlund J.T. et al. Rationale and design of Total Therapy Study XV for newly diagnosed childhood acute lymphoblastic leukemia. Ann Hematol 2004; 83 (Suppl 1): 124–6.
66. Исаева Е.А., Жаринов В.С., Наседкина Т.В. и др. Анализ хромосомных аберраций и их прогностическое значение при остром лимфобластном лейкозе у детей. Вопр гематол онкол иммунопатол педиатр 2003; 2 (4): 59–66.
67. Rabbits T.H. Chromosomal translocations in human cancer. Nature 1994; 372: 143–9.
68. Rubnitz J.E., Look A.T. Molecular basis of leukemogenesis. Curr Opin Hematol 1998;5: 264–70.
69. Митяева О.Н., Наседкина Т.В., Жаринов В.С. и др. Анализ хромосомных транслокаций с участием гена MLL методом гибридизации с олигонуклеотидными микрочипами. Мол биол 2004; 38(3): 376–82.
70. Gra O.A., Sidorova J.V., Nikitin E.A. et al. Analysis of TCR gamma gene rearrangements using oligonucleotide microchip: a novel approach for the determination of T-cell clonality. (in press).
71. Nebert D.W. Polymorphisms in drug-metabolizing enzymes: what is their clinical relevance and why do they exist? Am J Hum Genet 1997; 60: 265–71.
72. Nebert D.W., McKinnon R.A., Puga A. Human drug-metabolizing enzyme polymorphisms: effects on risk of toxicity and cancer. DNA Cell Biol 1996; 15: 273–80.
73. Elion G.B. The purine path to chemotherapy. Science 1989; 244: 441–7.
74. Weinshilboum R.M., Sladek S.L. Mercaptopurine pharmacogenetics: monogenic inheritance of erythrocyte thiopurine methyltransferase activity. Am J Hum Genet 1980; 32: 651–62.
75. McLeod H.L., Lin J.S., Scott M.C. et al. Thiopurine methyltransferase activity in American white subjects and black subjects. Clin Pharmacol Ther 1994; 55: 15–20.
76. Roberts W.M., Estrov Z., Kitchingam G.R. et al. The clinical significance o